Download

1 / 69
690 likes | 874 Views
第八章 芳烃 ( Aromatic Hydrocarbon ). ◇ 芳烃是芳香(族)化合物的母体,最简单的芳烃是 苯( benzene )。. ◇ “ 芳香化合物 (aromatic compounds) ” 概念的演变: 芳烃是芳香 ( 族 ) 化合物的母体,最简单的芳烃是苯 (benzene) 。. 一、苯的结构. 1. 凯库勒式 ◇ 苯的凯库勒结构式的导出 1865 年, Kekul é ( 德国 ). ◇ 苯的凯库勒结构式不能代表苯的真实结构 ◎ 原因之一: 氢化热

E N D
◇芳烃是芳香(族)化合物的母体,最简单的芳烃是苯(benzene)。◇芳烃是芳香(族)化合物的母体,最简单的芳烃是苯(benzene)。 ◇“芳香化合物(aromatic compounds)”概念的演变: • 芳烃是芳香(族)化合物的母体,最简单的芳烃是苯(benzene)。
一、苯的结构 1.凯库勒式 ◇苯的凯库勒结构式的导出 1865年,Kekulé(德国)
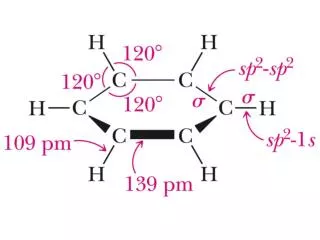
◇苯的凯库勒结构式不能代表苯的真实结构 ◎原因之一:氢化热 即苯的真实结构比kekulé的环已三烯式稳定358.5-208.5=150 kJ/mol 故苯中无三个双键存在。 ◎原因之二:苯的Kekulé式不能说明苯的结构的实验测定结果。 按kekulé式,苯中有单双键交替,键长不等,为不规则六边形,而实测结果是,苯中6个碳碳键等长(139.7pm),<HCC= <CCC=120° 那么苯的真实结构到底如何?或者说,究竟怎样描述苯的真实结构?
2.苯的分子轨道模型 ◇分子骨架 ◇闭合共轭体系:各C所余的P轨道与平面垂直,相互平行,相邻的P轨道相互侧面重叠,形成六电子六个碳的闭合共轭体系。
HMO结果为: 基态时6个P电子均处成键轨道 苯分子中6个π电子的总能量=2×(α+2β)+4×(α+β) =6 α+8β
该6个π电子分别处于三个孤立π轨道中的总能量=6×(α+β)该6个π电子分别处于三个孤立π轨道中的总能量=6×(α+β) = 6α+6β 显然(6α+8β)-(6α+6β)=2β<0 ∴苯为很稳定的体系,其中无孤立π键存在 因π电子离域,故键长均等=139.7pm 134pm(孤立C=C)<139.7pm(苯中C-C键长)<154pm(烷中C-C单键) 即苯中C-C键介于单键和双键之间 3.苯的共振式、共振能、苯结构表示法 ◇苯的结构也可用共振式表示: 意义:苯是两个经典结构(凯库勒式)的共振杂化体。
◇共振能(RE,Resonace Energy) ◎定义:RE=经典结构式的能量-实际分子的能量 ◎意义:表示经典结构式的“共振”所起的稳定作用的大小 ◎估算法: 例:可用氢化热估算苯的RE 即,由于共振,使 比经典结构环已三烯 稳定150.0kJ/mol
◇苯结构表示法: ◎正六边形内加一个圆圈: 强调了苯中π电子云的平均分布,但没说明π电子数 ◎正六边形加三个双键 但这不是环已三烯,而是共振式的简写
二、苯衍生物的异构和命名 ◇苯的一元衍生物(C6H5A)只有一种 命名法
◇苯的二元取代物(C6H4AA或C6H4AB)有三种异构体 相对位置表示法: 邻、o(ortho),1,2- 间、m(meta),1,3- 对、p(para),1,4- 例: ◇苯的三元取代物且三个取代基相同时(C6H3AAA)有三种异构体 相对位置表示法: 连、vic(vicinal),1,2,3- 偏、unsym(unsymmetrical),1,2,4- 均、sym(symmetrical),1,3,5-
例: 问题:二溴苯有三种异构体,其熔点分别为87.33℃、7.1℃和-7℃。从熔 点为87.33℃的二溴苯只能得到一种硝基化合物C6H3Br2NO2,从熔点为7.1℃ 的二溴苯只能得到两种一硝基化合物,从熔点为-7℃的二溴苯可以得到三 种一硝基化合物。试推测三种二溴苯的构造。 答案:
三、苯环上的亲电取代反应 ◇苯的芳香性 易取代 难加成和氧化 碳环异常稳定 取代 亲电取代 • 卤化(-X) • 硝化(-NO2) • 磺化(-SO3H) • 酰基化(-COR) • 烷基化(-R) 亲核取代 自由基取代
能线图为: 1.卤化反应(Halogenation) ◇试剂(卤化剂):Cl2,Br2 ◇催化剂:Fe屑或FeX3 ◇亲电试剂:X+
◇例: 2.硝化反应(Nitration) ◇试剂(硝化剂):混酸(浓HNO3+浓H2SO4) ◇催化剂:H2SO4(含于试剂中) ◇亲电试剂:NO2+(硝鎓离子或硝基正离子)
◇例: ◇机理: 3.磺化反应(Sulfonation) ◇试剂(磺化剂): 发烟硫酸-含SO3的浓硫酸(快) 浓硫酸(很慢) 还可用SO3,ClSO3H ◇催化剂:无 ◇亲电试剂:
◇例: ◇特征:磺化反应可逆 如:
4.Friedel-Crafts反应 ◇1877年,由巴黎大学的法国化学家Friedel和美国化学家Crafts两人 首先发现,是有机合成中最有用的反应之一。 (1)酰化反应(Acylation) ◇试剂(酰基化剂): ◇催化剂:无水AlCl3 ◇亲电试剂:
◇例: (2)烷化反应(Alkylation) ◇试剂(烷基化试剂):卤烷、烯烃、醇 ◇催化剂:AlCl3,FeCl3,BF3等Lewis酸 ◇亲电试剂:R+(烷基正离子)
如: ◇例子: ◇特征: ◎多烷基化产物的生成 由于烷基使芳烃活化,故烷基苯起烷化反应的速度比苯更快:
为减少二烷基化及多烷基化产物的生成,必须使用大量的苯。为减少二烷基化及多烷基化产物的生成,必须使用大量的苯。 ◎有重排产物生成 例: 解释:亲电试剂为R+,而R+可重排生成更稳定的R+。
四、苯环上取代反应的定位规律 1.定位规律070531 ◇比较三者的硝化反应:
◇结论: ◎亲电取代反应活性: 即-CH3使苯环活化(致活),-NO2使苯环钝化(致钝)
◎苯环上原有的取代基对新导入的取代基有定位作用 (orientation),即当苯环上已有一个取代基时,第二个取代基主要进入苯环的位置由原有的第一个取代基所决定。 原有-CH3 -NO2主要进入邻、对位 -CH3为邻对位定位基 原有-NO2 -NO2主要进入间位 -NO2为间位定位基 或 Activating Groups Ortho-Para Directors Deactivating Groups Meta Directors ◇定位规律:当一元取代苯通过亲电取代反应引入第二个取代基E时,由G 决定E进入苯环的位置及苯环发生该亲电取代反应的相对活性 ◇G的分类:两大类 ◎第一类:邻对位定位基 指令E主要进入其邻对位:
▲又分为: 使苯环活化的邻对位定位基:如:-N(CH3)2,-NH2,-OH,-OCH3,-NHCOCH3,-C6H5,-CH3等 使苯环钝化的邻对位定位基:指卤素:-F,-Cl,-Br, -I ▲结构特点: G中和苯环直接相连的原子外层均有未成对电子 如: 可与苯环大π键发生δ-π超共轭(如-CH3) ◎第二类:间位定位基 指令E主要进入其间位: 使苯环钝化 例:-N+(CH3)3,-NO2,-CN,-SO3H,-CHO,-COCH3, -COOH,-COOR,-CF3等
▲结构特点: 和苯环直接相连的原子上带有正电荷 以重键和一个电负性较大的原子相结合再与苯环连接 2.定位规律的理论根据 ◇定位规律是一个经验规律,1895年由Holleman总结出。可用共振论(RT)解释之。 ◇从芳烃亲电取代历程可知,σ络合物的生成是决速步骤,因而σ络合物的稳定性决定整个亲电取代反应的快慢。
若某种σ络合物(C+)较稳定 反应所需的活化能较小 反应速度较快 相应的产物在总产物中所占的份额较多 G为该种位置定位。 例:解释-CH3为邻、对位定位基 甲苯在亲电取代反应中生成的三种σ络合物的结构可用共振式表示:
当亲电试剂E+进攻G的邻或对位时,生成的碳正离子(或σ络合物)的经典结构式中有一个是叔碳正离子,其余两个是仲碳正离子。当亲电试剂E+进攻G的邻或对位时,生成的碳正离子(或σ络合物)的经典结构式中有一个是叔碳正离子,其余两个是仲碳正离子。
当E+进攻G的间位时,三个经典结构式都为仲碳正离子当E+进攻G的间位时,三个经典结构式都为仲碳正离子 ∴稳定性: (1)、(2)>(3) 取代产物中以邻位和对位异构体为主 即甲基为邻、对位定位基 或者说: (1)和(2)的共振式中都有一个经典结构为 C+CH3,CH3给电子,使之较稳定,(3)中无此情况,故σ络合物(1)、(2)均比(3)稳定。 3.二取代苯的取代反应 (1)两取代基定位效应一致时,其作用具加合性
例: (2)两取代基定位效应矛盾时 同类基,由定位效应强者决定 不同类基,一般由第一类基决定 例:
例:用箭头表示下列化合物在硝化反应中硝基所占的位置(主要产物)例:用箭头表示下列化合物在硝化反应中硝基所占的位置(主要产物)
4.分速度因素(partial rate factor) ◇定义:一元取代基C6H5R分子中苯环上某一特定位置与苯分子中一个位置相比较,起取代反应的相对速度,称为这一位置的分速度因素。 ◇物理意义:定量反映出取代苯不同位置(邻、间、对)起亲电取代反应的相对活性。 ◇计算式:fz=6/y×kr×x fz-z位置的分速度因素 z-邻(o),对(p)或间(m)位 y-z位置的数目 kr-取代基与苯的相对取代反应速度常数
kr可用竞争法测定 如:将等摩尔的甲苯和苯与少量硝酸在乙酐溶液中,30℃下反应,生成的产物用色谱法分析,所得硝基苯与硝基甲苯分数之比为1:27 则 (∵生成的硝基甲苯与硝基苯的量与甲苯和苯起硝化反应的速 度成正比) X-z位置异构产物的分数 ◇例子:甲苯硝化(CH3COONO2,0℃)时的异构体生成比例分别为:邻位61.4%,间位1.6%,对位37.0%,kr=27.0,求各位置的分速度因素。 解:
说明: f0、fp、fm均大于1,表示甲苯的反应活性比苯大(或甲基使苯环活化) 底物选择性(Substrate Selectivity) 而f0、fp>>fm,表示甲苯使邻对位活化的作用大于间位,即CH3为邻对位定位基 位置选择性(Position Selectivity) 5.定位规律的应用 ◇巧妙地利用取代基的定位规律,合理地确定取代基进入苯环的先后次序可有效地合成芳族化合物。 ◇例1:
五、烷基苯的反应 1.侧链卤化 (1)侧链氯化 控制Cl2用量可使反应停留在苄氯阶段,因苄基自由基 中有p-π共轭,较稳定(其机理为自由基取代)
(2)侧链溴化 2.氧化 ◇有α-H的芳烃侧链(烷基)易被氧化成-COOH ◇氧化剂 KMnO4 + H2SO4 NaCr2O7 + H2SO4 稀HNO3等
3.催化加氢 难进行 例: 4.Birch还原 例:
六、多环芳烃 1.多苯代脂烃 ◇定义:链烃分子中两个以上的H被苯基取代生成的化合物。 例: (1)三苯甲烷 ◇制备:
◇反应: ◇三苯氯甲烷 反应: 三苯甲基正离子(C6H5)3C+的正电荷可分散到三个苯环中去,使其 稳定性大幅度提高。 C+的稳定性为:(C6H5)3C+> (C6H5)2CH+>R3C+>R2CH+≈C6H5CH2+ ≈CH2=CH-CH2+>RCH2+>CH3+
(2)三苯甲基自由基 ◇1900年,Gomberg用实验证明自由基可独立存在:
◇三苯自由基稳定的原因:p-π共轭 • 其未配对电子可分散在10个C上 (3)三苯甲烷染料或指示剂
3.稠环芳烃 ◇定义:两个苯环共有两个C 例: (1)萘 ◇物性:无色晶体,mp=80.55℃,易升华 ◇表示: