Download

1 / 46
520 likes | 896 Views
The Protein Folding Problem When will it be solved?. Dill et al, 2007. What is protein folding ? Why is it a problem ? What are some approaches to understanding it? How far have we come? What does the future hold?.

E N D
The Protein Folding ProblemWhen will it be solved? Dill et al, 2007
What is protein folding? Why is it a problem? What are some approaches to understanding it? How far have we come? What does the future hold?
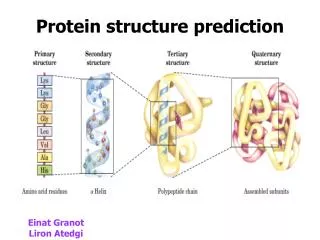
Protein folding is how an amino acid sequence (a polypeptide) folds into its native structure.A native structure is the functional form of a protein.
The overall protein folding problem is to understand the relationship between amino-acid sequence and protein structure.
The protein design problem is to synthesize a stable amino-acid sequence towards a target conformation.
Understanding how inter-atomic forces contribute to a protein’s native structure.What is the driving force behind protein folding? Does understanding the problem have other levels of applicability? • Prediction of native structure from a given amino acid sequence.Can we input a polypeptide sequence and output the ‘correct’ protein? How accurate would this simulation be? • The kinematic question of folding speedJust how do proteins fold so fast? Can we attain this speed and accuracy with synthetic, de novo proteins?
Before mid-1980s, overall folding process was seen as sum of small, local interactions.Like hydrogen-bonds, van der Waal’s interactions, ion pairs. • Statistical mechanical modeling after mid-80s changed this view. • Big component is reducing exposed hydrophobic sidechains. I.e., non-local interactions are the ‘driving force’. • Folding process is distributed locally and non-locally • Free Energy Equation • Effects of cytosol cannot be ignored. • Composition (solvent, other macromolecules, pH) • Temperature
Design of foldamersSynthetic molecules which mimic folding ability of proteins (e.g. peptoids in pharmaceutics) • Design of lung surfactant replacements • Cytomegalovirus inhibitors • Antimicrobials • siRNA delivery agents • Synthetic proteins from “broadened alphabets”
Bioinformatics-based approach • Long-standing goal.Amino-acid sequence in→ 3-D Native Structure out • Makes drug discovery faster.Simulate drug interactions without costly studies. • Makes it cheaper.Replace experimental structural determination with accurate computer simulation.
CASP • Critical Assessment of Techniques for Structure Prediction (Moult, 1994) • Social experiment • Prediction of native state from amino-acid sequence alone • Approaches are homology modeling and protein threading
Physics-based approach • Use only the laws of Physics to model folding processes and resulting native structures. • Aim to not use statistical energy functions or secondary structure predictors.Like Homology Modeling, Protein Threading. • Now being combined with some database information.
Physics-based approach – Advantages • Can predict conformational changesInduced-fit theory: important in computational drug discovery • Predict conformational transitionsMaybe those based on environmental factors • Design synthetic proteinsFoldable polymers for non-biological backbones
Physics-based approach – Problems • Inaccuracies in “force-fields” • Really, really high computational requirementsAt least right now
Computational Cost One year to simulate folding of small proteinapproximately…
Recent Advances • 36-residue villin folded in 1μs • Explicit solvent, initially unfolded • Duan, Kollman (1998) • 4.5A RMSD • 20-residue Trp-cage folded in 92ns • Implicit solvent • Around 1A RMSD • Folding@Home folded villin to 1.7A RMSD
Root Mean Square Deviation If molecular orientation changes in arbitrary way, lRMSD or Least RMSD is used to find optimal alignment using the Kabsch Algorithm or Quaternions
Afinsen’s Paradigm (1957)All the information required to make a 3-D native structure is contained in the amino-acid sequence • Levinthal’s Paradox (1968)If a protein sampled all possible conformations, time to reach ‘correct’ one would be more than age of universe • Protein Sequence SpaceWith 20 amino acids as ‘alphabet’, how many theoretical proteins are possible? What about evolution? • Baldwin ConjectureUnderstanding protein folding can lead us to devise better algorithms to predict native structures from amino-acid sequences
Folding speed • Two decades ago, could not measure anything faster than few milliseconds. • Technology exists now. • Laser temperature-jump methods • Mutational methods to identify amino-acids controlling folding speed • Förster resonance energy transfer (FRET) methods to ‘watch’ formation of contacts • Hydrogen-exchange methods to ‘see’ structural events • Extensive studies on protein modelsCytochrome c, barnase, chymotrypsin inhibitor 2
What we know • Folding does not happen in a single microscopic pathway • “Funnel-shaped” landscapes • Going from non-native state to native state is different for each conformation of same sequence • Folding processes are heterogenous • Observations see averages and not distributions, variations
What we don’t know • How do folding rates change with specific mutations? • How to characterize kinetic heterogeneity? • Single-molecule experiments • Master-equation theories
Question • How to design simulations which can arrive at native state faster and more accurately than Monte Carlo or molecular dynamics?Need to know microscopic folding routes
A possible answer • Zipping and Assembly (ZA) • Proteins do not reach all their degrees of freedom at the same time • Fold over a range of timescales. • Fast timescale (nano to pico): Small peptide pieces explore conformations independently. • Formed local structure “zips”, includes more surrounding chain. • Further assembly on slower timescale.
Does it work? • ZA speeds up conformational searching. • Physics-only models can find approximately correct folds for 25-75 monomers. • Ozkan et al, 2006 • Used AMBER96 force-field, implicit solvent • Tested 9 proteins from PDB • Eight were within 2.2A (avg.) RMSD • Gives a good overall sense of folding mechanism
Difficulty Can we know the conformations the overall protein does not search?Important in understanding proteopathies and hence drug design.
Sophisticated problem • Protein-protein interactions, cofactors, multi-domain protein behavior, cytosolic interactions and effects unknown . • But some headway is being made • Small proteins’ native structures and folding codes are being determined accurately • What we know is sufficient to design new proteins and polymers (foldamers) • Good contributions to novel drug discovery and proteomics • Good sense of Levinthal’s optimization puzzle
Structure is related to function • Resulting protein is rendered biologically, functionally inactive • Simpler forms can be degraded by cell machinery • Amyloid accumulation (Proteopathies)Alzheimer’s, Parkinson’s • Can re-fold other normal proteins (Prions)Creutzfeld-Jakob Disease
Sources • http://www3.interscience.wiley.com/journal/66000862/abstract?CRETRY=1&SRETRY=0 • http://opa.faseb.org/pdf/protfold.pdf • http://www.biozentrum.unibas.ch/~schwede/Teaching/BixII-SS05/FR-HM.pdf • http://dasher.wustl.edu/bio5476/reading/curropstrbio-14-76-04.pdf • http://arxiv.org/ftp/q-bio/papers/0402/0402039.pdf • http://www.biostat.jhsph.edu/~iruczins/presentations/ruczinski.04.04.retreat.pdf