Download

1 / 2
30 likes | 51 Views
In order to assess the biological function of proteins and their modifications for understanding signaling mechanisms within cells as well as specific biomarkers to disease, it is important to study the change of proteome under different experimental conditions. The emerging technology of mass spectrometry-based quantitative proteomics provides a powerful tool to quantitatively and systematically assess quantitative differences in protein profiles of distinct samples and is increasingly becoming a significant component of biomedical and clinical research.

E N D
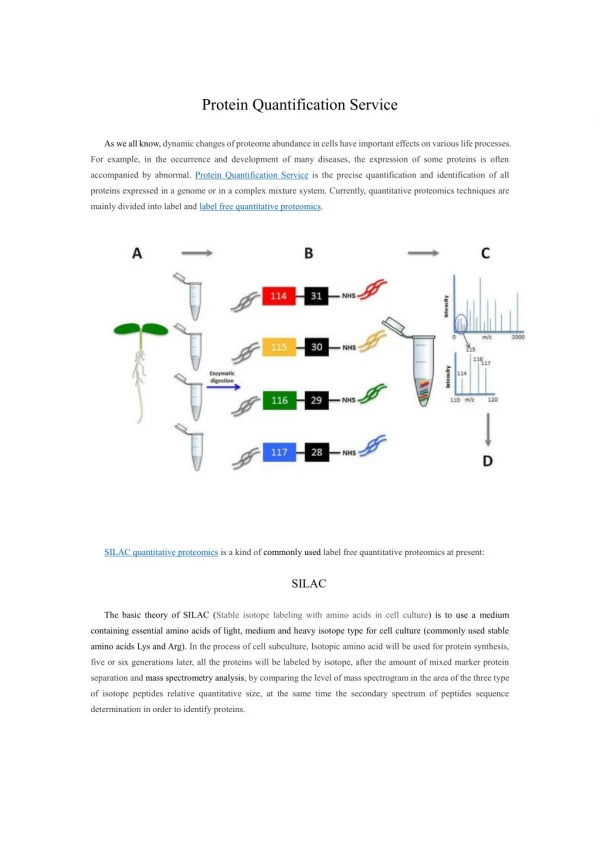
Protein Quantification In order to assess the biological function of proteins and their modifications for understanding signaling mechanisms within cells as well as specific biomarkers to disease, it is important to study the change of proteome under different experimental conditions. The emerging technology of mass spectrometry-based quantitative proteomics provides a powerful tool to quantitatively and systematically assess quantitative differences in protein profiles of distinct samples and is increasingly becoming a significant component of biomedical and clinical research. Protein Quantification Service Discovery-based quantitative proteomics compares the proteome of a diseased sample versus normal at a global scale, and has been widely used to study various human diseases with the goal to identify biomarkers and/or reveal the pathogenesis of diseases. Methods for acquiring quantitative proteomics data are continually developing with very accurate stable isotope labeling (SIL) and label-free approaches. SIL provides chemically equivalent but isotopically different interior standards for each peptide/protein for direct comparison of mass spectral signal intensities that represent relative abundance Common SIL strategies include protein and peptide level labeling strategies such as: SILAC: stable isotope labeling of amino acids in cell culture,a global method whereby all translated proteins have isotope labels metabolically incorporated at selected amino acid residues; ICAT: isotope-coded affinity tags, a technique that labels cysteine residues at the protein level iTRAQ: multiplexed isobaric tags for relative and absolute quantification; GIST: global internal standard technology, a global post-digestion labeling method that labels primary amine groups (peptide N-terminus and lysine residues) ISIL: an extension of GIST called in-gel stable isotope labeling, a method that labels primary amine groups of proteins (protein N-terminus and lysine residues) directly from gel separated samples. Label-free: approaches to quantitative proteomics have gained prominence in recent years since no additional chemistry or sample preparation steps are required. Mass spectrometry-based approaches for targeted protein quantification are based on the concept of isotope dilution mass spectrometry techniques commonly used for the detection of small molecules. The approach is
comparable to modern advanced antibody-based methods, such as the multianalyte profiling platform (MAP), offering multiplexing detection capability with an excellent dynamic range. With years’ experience in advanced experiment equipment, Creative Proteomics can provide a variety of proteomics services to assist your scientific research, including: iTRAQ-based proteomics analysis service TMT-based proteomics analysis service SILAC-based proteomics analysis service Absolute quantification (AQUA) service Label-free quantification service Semi-quantitative proteomics analysis service Parallel Reaction Monitoring (PRM) SRM&MRM As one of the leading omics industry company, Creative Proteomics now is opening to provide a series of protein quantification services for our customers. Our service guarantees accurate and reliable results, at quick turnaround time. Please feel free to contact us and see how we can help to address your problems.