Download

1 / 41
530 likes | 2.03k Views
Histone Modifications. Seminar in Bioinformatics Saar Gershuni 2005. Background – DNA Packing. The DNA is packed in various levels of condensation in the nucleous (*10,000) Form of condensation – biological role Chromosomes, Euchromatin, heterochromatin, DNA strand. The Beads on a String.

E N D
Histone Modifications Seminar in Bioinformatics Saar Gershuni 2005
Background – DNA Packing • The DNA is packed in various levels of condensation in the nucleous (*10,000) • Form of condensation – biological role • Chromosomes, Euchromatin, heterochromatin, DNA strand
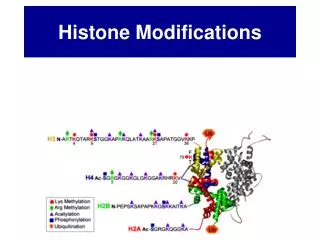
The Beads on a String • The Histones form the 11nm strand. • Are octamer build H3,H4,2HA,2HB monomers • 146-147 bp are wrapped around every histone core • The histone tail sequences account for 28% of the total amino acid content of the core histones “chromatin fiber folding: requirement for the Histone H4 n-terminal tail “, Benedetta Dorigo†, Thomas Schalch†, Kerstin Bystricky†, ‡ and timothy J. Richmond, journal of molecular biology 327, 1 , 14 march 2003 • Tail of histone h4 taken from cow
Chemical Review • Acetyl • Methyl • Phosphoryl • Ubiquitin
Histone Modifications • De/Acetylation • Methylation • Phosphorylation • Ubiquitination • ADP-Rybosilation • Swi/Snf complex, which, in vitro, uses the energy of ATP hydrolysis to disrupt histone-DNA interactions
Histone Modifications - Role • Transcription – Acetylation/Methylation • DNA repair – H2A -Phosphorilation • Mitosis – chromosomal arrengement • Chromatin assembly – DNA replication
Figure 1. Sites of post-translational modificationson the histone tails Yi Zhang et al. Genes Dev. 2001; 15: 2343-2360
Examples of Biological Role of Histone Modifications Acetylation –Lisine • Transcription – loosening the strand • Replication – the positioning of histones • Gcn5 – h3k14
Examples of Biological Role of Histone Modifications Phosphorylation – serine, threonine (scienceweek) • Chromosomal condensation – H1,H3 • Rsk-2, an H3 kinase, Coffin Lowry syndrome • Transcription regulation - drosophila sex chromosomes serine 10 H3 in concert with H4K16
Coffin Lowry Syndrome Coffin-Lowry syndrome is a rare genetic disorder characterized by mental retardation; abnormalities of the head and facial area; large, soft hands with short, thin (tapered) fingers; short stature; and/or various skeletal abnormalities. Characteristic facial features may include an underdeveloped upper jawbone, an abnormally prominent brow, downslanting eyelid folds, widely spaced eyes, large ears, and/or unusually thick eyebrows. Skeletal abnormalities may include abnormal front-to-back and side-to-side curvature of the spine and unusual prominence of the breastbone. Coffin-Lowry syndrome is caused by mutations in the RSK2 gene and is inherited as an X-linked dominant genetic trait. Males are usually more severely affected than females.
Examples of Biological Role of Histone Modifications Methylation – Arginine, Lisine • Less studied, enzymology – not known • Can be mono-, bi-, tri- methylated • Transcription regulation - CARM1, arginine-specific, histone h3-selective methyltransferase activity, coactivator, with p160 family
Figure 2. Chemistry of arginine and lysine methylation Yi Zhang et al. Genes Dev. 2001; 15: 2343-2360
Histone Code • Code - a system of signals or symbols for communication (webster meriam online) • Requirements from a code: • Consistent • Combinatorial (Kurdistany & Grunstein, 2003)
Mapping Global Histone Acetylation Patternsto Gene ExpressionSiavash K. Kurdistani, Saeed Tavazoie,and Michael Grunstein
Introduction • The mechanism by which histone de/acetylation affect transcription involve two pathways: • By altering the folding properties of the chromatin fiber • By providing binding surface for recruitment of other elements • To date (6/2004) there is no evidence for consistent patterns of de/acetylation from gene to gene or for the combinatorial use of histone modification sites
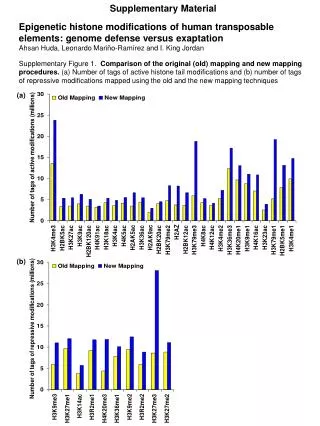
The Experiment – Data Collection & Methods • Chromatin was extracted from YDS2 exponentially growing • Using chip and DNA microarrays levels of modification was determined • 11 sites of acetylation were examined – H4k8,12,16 H3k9,14,18,23,27 H2ak7,h2bk11,16
The Experiment - Methods • Two Microarrays: 6700 IGR, 6200< ORF • 2-4 repetitions • Normalization by the ration of total intensities. • Coefficient of variation < 0.5 between replicate experiment were counted • End up with 2206 IGR, 2403 ORF • Data were again normalized over the 11 sites per histone
Results – Correlation With Gene Expression These results – though significant might be artificially low due to technicalities.
Results – coexpression of Clusters Problem: what is the expression level of randomly selected cluster of genes? The study also checked expression levels at different stress conditions (255) – correlation was found in 6 IGR’s and 13 ORF’s
Results – Clusters Biological Relevance • Annotations for all the genes in a cluster were taken from MIPS, GO, MDS • 12/53 IGR’s, 13/68 ORF’s – with significant results • Motif search using AlignACE algorithm found 102 of 29/53 IGR’s, 110 of 34/68 ORF’s
Does the Modifications Constitute a Code? • The authors believe that the answer is no because: • The total number of modifications does not contain more information than the sum of individual modification. • Problem: it has been shown to be combinatorial – bdf1 in vitro preference for tetra acetylated H4.
Problems • Cutting the chromatin fiber was done using sonicator bath thus creating various size of fiber – with various number of nucleosome – problem with measuring acetylation levels. • Microarrays are 1kb in length can contain up to 5 nucleosomes.
Problems • Normalization – step 1 – average number of 1 through all the genome.Step 2 – normalizing groups of 11 lysines in each and every locus (=0, var=1) • All the problems relates with k means algorithm, AlignACE, and gene expression data
Genomic Maps and Comparative Analysis of Histone Modifications in Human and Mouse Bradley E. Bernstein, Michael Kamal, Kerstin Lindblad-Toh, Stefan Bekiranov, Dione K. Bailey, Dana J. Huebert,Scott McMahon, Elinor K. Karlsson, Edward J. Kulbokas III, Thomas R. Gingeras, Stuart L. Schreiber, and Eric S. Lander
The Experiment • Large scale study of histone modifications (methylation, acetylation) patterns in human and mouse cells • Methods: ChIP, RTPCR – for validating the data, tiling oligonucleotide arrays – 35 bp intervals • Focus: chromosomes 21,22, (H3K4 di/trimethylation and H3K9,14 acetylation) and cytokine cluster, IL4 Receptor, and Hox clusters (H3K4 dimethylation)
Results – Raw Data • 90%< correlation between methylated H3K4, and acetylated H3K9,14 • Di/Trimethyl – gene start
Conservation of Modification pettern Between Human and Mouse • For this purpose the IL4R methylation analysis were preformed • 55% conservation in human, 68% conservation in mouse, (*7 than random) with no correlation with sequence conservation
Hox Clusters • A group of linked regulatory homeobox genes that are involved in patterning the animal body axis during development. Homeobox genes are defined as those that contain an 180-base-pair sequence that encodes a DNA-binding helix–lturn–helix motif (a homeodomain).(Nature) • The remaining orthologous regions between human and mouse
Methylation Patterns in Hox Clusters • Completely unique. • Contain huge methylated regions encompass multiple genes • Evolutionary conserved (human-mouse) • Methylation correlates with expression both in ORF’s and IGR’s unlike IL4R
Problems • The method of creating the lysate is still sonication. • No relation with genes functionality, cell cycle phase – can change nucleosome concentration
What Is It Good for? • Genome wide understanding of chromatin role (structure, functionality( in biology • The use of all the methods we learned • Improve our understanding regarding various mechanisms and processes within the nucleous • Develop new bioinformatic and biological methods for research (advanced ChIP technics, combination with tiling microarrays, and data analyzing tools – normalizations, intergrated tools)
Where Do We Go Next? • Characterization of lysine 56 of histone H3 as an acetylation site in Saccharomyces cerevisiae.Ozdemir A, Spicuglia S, Lasonder E, Vermeulen M, Campsteijn C, Stunnenberg HG, Logie C. • Epigenomic mapping in Arabidopsis using tiling microarrays.Martienssen RA, Doerge RW, Colot V. • The epigenetic breakdown of cancer cells: from DNA methylation to histone modifications.Ballestar E, Esteller M.