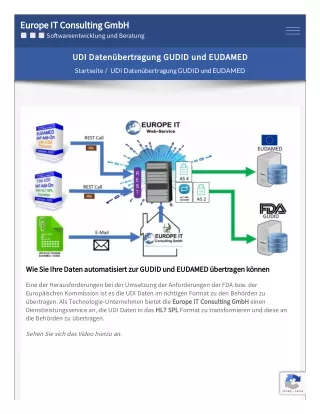
Download

1 / 82
830 likes | 1.82k Views
It takes a decade on average for an experimental drug to travel from lab to medicine chest. ... Greece 3. Guatemala 2. Hong Kong 6. Hungary 13. Ireland 1. Israel 5. Italy ...

E N D
Slide 1:FDA Expectations of Clinical Trials and Investigators Joseph P. Salewski Deputy Director Division of Scientific Investigations (DSI) Center for Drug Evaluation and Research National Labor College, November 8-11, 2010
2Slide 2:Center for Drug Evaluation and Research
Slide 3:Division of Scientific Investigations
Informatics and Infrastructure Enforcement Policy
4Slide 4:Presentation Outline
Overview of BIMO DSI�s Role & Authority in BIMO Program GCP Guidance vs Regulations The Clinical Investigator Role & Responsibility Selection and Auditing CI Sites PDUFA vs For Cause Post Inspectional Procedures/DSI Actions Consequences Sponsors
5Slide 5: FDA�s Bioresearch Monitoring Program - A comprehensive program of on-site inspections and data audits designed to monitor all aspects of the conduct and reporting of FDA regulated research.
FDA�s BIMO Program
6Slide 6:BIMO Program Objectives
To verify the quality and integrity of research data To ensure that the rights and welfare of human research subjects are protected
7Slide 7:Presentation Outline
Overview of BIMO DSI�s Role & Authority in BIMO Program GCP Guidance vs Regulations The Clinical Investigator Role & Responsibility Selection and Auditing CI Sites PDUFA vs For Cause Post Inspectional Procedures/DSI Actions Consequences Sponsors
8Slide 8:DSI/CDER�s BIMO Program Responsibilities
Ensure adherence to applicable regulations with respect to: Good Laboratory Practice (GLP) In vivo Bioequivalence Good Clinical Practice (GCP) Institutional Review Boards Clinical Investigators Sponsor-Monitors, CROs
9Slide 9: What does DSI do?
Assigns and Performs inspections through the Office of Regulatory Affairs (ORA) to verify data submitted to FDA and ensure the protection of the rights and welfare of human research subjects Investigates allegations of regulatory non-compliance Provides a scientific and medical review of Establishment Inspection Reports (EIRs) generated by ORA Makes recommendations regarding data reliability to Review Divisions Takes appropriate regulatory action
10Slide 10:Regulatory Authority to Conduct Inspections/Audits
21 CFR 312.68 �An investigator shall upon request from any properly authorized officer or employee of FDA, at reasonable times, permit such officer or employee to have access to, and copy and verify any records or reports made by the investigator��
11Slide 11:FDA/CDER GCP Regulations
12Slide 12:Drug Development and Review Process It takes a decade on average for an experimental drug to travel from lab to medicine chest. Only five in 4,000 compounds screened in preclinical testing make it to human testing. One of these five tested in people is approved.
YEARS 1 2 Preclinical Testing Test Population Laboratory and Animal Studies PURPOSE Assess toxicity and biological activity % of all new drugs that pass FILE IND Phase I Phase II Phase III 3 4 5 6 7 20 to 80 Healthy Volunteers 100 to 300 Subject Volunteers 1,000 to 3,000 Subject Volunteers 8 Determine Acute Toxicity and Dosage Evaluate effectiveness. Look for Side effects. Verify effectiveness, monitor adverse reactions from Cumulative dosing and delayed Toxicity Expedited Review: Phases II and III combined to shorten approval process on new medicines for serious & life-threatening diseases. 70% of INDs 33% of INDs 27% of INDs FILE NDA 9 10 Review usually takes about - 1 year Post-marketing safety monitoring Distribution Education 20% of INDs FDA Approval
Slide 13:When Do I Need an IND?
CFR 312.2 (b) Exemptions The clinical investigation of a drug product that is lawfully marketed in the United States is exempt from the requirements of this part if all of the following apply: (i) The investigation is not intended to be reported to FDA as a well-controlled study in support of a new indication for use or not intended to be used to support any other significant change in the labeling for the drug; (ii) If the drug that is undergoing investigation is lawfully marketed as a prescription drug product, the investigation is not intended to support a significant change in the advertising for the drug;
Slide 14:When Do I Need an IND?
CFR 312.2 (b) Exemptions (continued) (iii) The investigation does not involve a route of administration or dosage level or use in a patient population or other factor that significantly increases the risks (or decreases the acceptability of the risks) associated with the use of the drug product; (iv) The investigation is conducted in compliance with the requirements for institutional review set forth in part 56 and with the requirements for informed consent set forth in part 50; and (v) The investigation is conducted in compliance with the requirements of CFR 312.7 (Promotion and charging for investigational drugs).
Slide 15:Who Do I Contact to Find Out if I Need an IND?
Contacts: Drugs: Barry Poole (301) 796-3145 Biologics: Robert Yetter (301) 827-0373 Devices: IDE Staff (240) 276-0125 Foods: David Hattan (202) 436-1293 Call, and then submit the request in writing (FAX or e-mail). FDA plans to issue a guidance for this question.
16Slide 16:Presentation Outline
Overview of BIMO DSI�s Role & Authority in BIMO Program GCP Guidance vs Regulations The Clinical Investigator Role & Responsibility Selection and Auditing CI Sites PDUFA vs For Cause Post Inspectional Procedures/DSI Actions Consequences Sponsors
17Slide 17:GCP Regulations
FDA has regulations governing the approval, conduct, review and reporting of clinical research intended for submission 21 CFR 312: IND regulations (rev. 2002) 21 CFR 50: Informed consent (rev. 2001) 21 CFR 54: Financial Disclosure 21 CFR 56: IRB (rev. 2002) 21 CFR 314: NDA regulations (rev. 2002) These are legally enforceable requirements.
18Slide 18:FDA �Guidance� Documents
Agency�s current thinking on GCP Not binding on FDA or the public An alternative approach may be used if such approach complies with the relevant statute or regulations
19Slide 19:GCP Guidance Documents
FDA has guidance documents which are not legal requirements but state acceptable standards Guidance for Industry, Investigators, & Reviewers � Exploratory IND studies (Jan. 2006) Guidance for Clinical Trial Sponsors: Establishment and Operation of Clinical Trial Data Monitoring Committees (March 2006)
20Slide 20:Regulation vs. Guidance
21Slide 21:Presentation Outline
Overview of BIMO DSI�s Role & Authority in BIMO Program GCP Guidance vs Regulations The Clinical Investigator Role & Responsibility Selection and Auditing CI Sites PDUFA vs For Cause Post Inspectional Procedures/DSI Actions Consequences Sponsors
22Slide 22:FDA Expectations of Clinical Investigators
Adherence to Code of Federal Regulations Knowledge of Clinical Investigator regulations Understanding Clinical Investigator responsibilities
Slide 23:Statement of Investigator 21 CFR Part 312
Form FDA 1572 Identifies the Clinical Investigator and the Sub-investigators (e.g., research fellows, residents, associates) who will be assisting the investigator in the conduct of the investigation
24Slide 24:Who is Listed on the 1572?
The investigator must sign the 1572 Item 6: Names of sub-investigators
25Slide 25:Definition
Investigator: an individual who actually conducts an investigation (under whose immediate direction the drug is administered or dispensed to a subject) Sub-investigator: any other individual member of the �study team�
26Slide 26:General Clinical Investigator Responsibilities
Ensuring that an investigation is conducted according to the Signed investigator statement (Form 1572) Investigational plan Applicable regulations Control of drugs under investigation Ensuring that informed consent is adequately obtained according to 21 CFR 50 Ensuring IRB review, approval and reporting requirements are met IAW 21 CFR 56
Slide 27:Investigator Responsibilities*
Follow the current protocol Personally conduct or supervise investigation(s) Ensure that all persons assisting in conduct of studies are informed of their obligations. Ensure informed consent (21 CFR 50) and IRB review, approval , and reporting (21 CFR 56) requirements are met. Obtain the informed consent of each human subject to whom the drug is administered. *(Form FDA 1572: #9. Commitments)
Slide 28:Investigator Responsibilities cont*
Notify the sponsor before making changes in the protocol. Notify the IRB and obtain IRB approval before making changes in the protocol. Report adverse events to the sponsor. Maintain adequate and accurate records. Make records available for inspection. Comply with all other requirements in 21 CFR 312 Report Financial Interests to the Sponsor (21 CFR 312) *(Form FDA 1572: #9. Commitments)
Slide 29:What Investigators Should Not Do
revise the protocol without obtaining the sponsor�s written concurrence neglect to submit the revised protocol to IRB for approval forget to obtain written informed consent and provide oral explanation of the study forget to update consent forms to reflect changes in the protocol
Slide 30:What Investigators Should Not Do
over-delegate to non-physicians (e.g., diagnosis that qualifies/determines eligibility for entry into the study) erase, white-out or obliterate original data entry either in CRFs or medical charts accept suggested changes to study data without checking the source documents or without justification for such changes
Slide 31:What Investigators Should Not Do
backdate the consent forms and signatures forget to obtain IRB approval of consent form revisions permit changes to study data without the investigator�s concurrence, especially after the investigator has �signed-off� the completed CRF blame anyone for inaccuracies in the CRFs
Slide 32:What Investigators Should Not Do
create fake records or patients by using demographic data or using blood, urine and tissue samples from other subjects alter patients� diaries to reflect a positive outcome use your staff as subjects in a study not having the condition(s) under investigation destroy study records
33Slide 33:Presentation Outline
Overview of BIMO DSI�s Role & Authority in BIMO Program GCP Guidance vs Regulations The Clinical Investigator Role & Responsibility Selection and Auditing CI Sites PDUFA vs For Cause Post Inspectional Procedures/DSI Actions Consequences Sponsors
34Slide 34:PDUFA vs For-Cause
For-Cause Inspections (Complaints) Based on complaints from any source Allegations that raise concerns regarding data integrity or the rights, welfare, and safety of study subjects have been compromised PDUFA-Related Inspections (NDA) FDA receives a Marketing Application Drug is typically an NME Pivotal studies not conducted under IND (Foreign) Data in support of application is only from foreign sites Also may be referred to as �Routine� Inspections
35Slide 35:Timing and Targets of BIMO Inspections
BIMO inspections can be conducted at any point in the drug development process Inspections during IND phase are generally �for cause = directed� Inspections during the NDA phase are generally �routine�, but can be �for cause� or �directed� May include Clinical Investigator (CI), Sponsor/Monitoring (S/M), Contract Research Organizations (CRO), Institutional Review Boards (IRB), Good Laboratory Practice (GLP), and Bioequivalence (BEq) inspection of FDA regulated research.
36Slide 36:PDUFA (Prescription Drug User Fee Act)- Related Inspections
Sponsor Submits New Drug Application to FDA FDA BIMO Inspection FDA Review Activities Marketing Approval OR Not
37Slide 37:PDUFA-Related: Selection of CIs
Site selection is a joint process: Review Divisions & DSI Site selection considerations: A specific safety concern at a particular site or sites based on review of AEs, SAEs, deaths, or discontinuations A specific efficacy concern based on review of site specific efficacy data Efficacy differential between sites Final outcome driven by a particular site or sites Efficacy outcome different than expected based on MOA of drug Specific concern for scientific misconduct at one or more particular sites based on review of financial disclosures, protocol violations, study discontinuations, safety and efficacy results
38Slide 38: Factors affecting CI Selection - PDUFA Inspections
Importance of the study Relevance to labeling/NDA Contribution/size/outliers Statistical impact of data from the site History of the clinical investigator Frequency and prior classification(s) Findings of previous inspection(s)
39Slide 39: Criteria for PDUFA International Inspections
Insufficient domestic data Only foreign data are submitted to support an application; Domestic and foreign data show conflicting results pertinent to decision-making; or Serious issues that need resolution, e.g., suspicion of fraud, scientific misconduct, significant human subject protection violations.
Slide 40:Clinical Investigator Inspections* CDER FY 2001-2009**
01 02 03 04 05 06 07 08 09 FY 283 277 *Based on inspection start date **FY09 to Date 368 351 366 408 369 407 458 1/5/10
Slide 41:Clinical Investigator Inspections �International* (CDER, FY 2002 � 2009**)
*Based on Inspection start date **FY09 to Date N=119 02 03 04 05 06 07 08 09 1/5/10
CDER-DSI International Clinical Investigator BIMO Inspections*: FY 2009** 21% 7% 4% 60% 7% n=119 1% 1/5/10 *Based on Inspection Start Date **FY09 to DateSlide 43:Total Number of Foreign Clinical Site Inspections by Country: 1980 � 2009
Algeria 1 Argentina 23 Australia 13 Austria 7 Bahamas 1 Belgium 29 Brazil 13 Canada 192 Chile 5 China 14 Columbia 1 Costa Rica 8 Croatia 12 Czechoslovakia 18 Denmark 18 Dominican Rep. 1 Estonia 5 Egypt 1 Finland 15 France 60 Gabon 1 Germany 63 Greece 3 Guatemala 2 Hong Kong 6 Hungary 13 Ireland 1 Israel 5 Italy 40 Japan 9 Kenya 2 Latvia 9 Lithuania 2 Malawi 1 Malaysia 1 Mexico 15 Netherlands 26 New Zealand 4 Nigeria 1 Norway 6 Panama 2 Peru 7 Philippines 2 Poland 33 Portugal 3 Republic of Korea 6 Romania 10 Russia 63 Slovenia 2 South Africa 32 Spain 16 Sweden 31 Switzerland 3 Taiwan 1 Thailand 7 U. K. 104 Ukraine 3 Venezuela 2 Zambia 1
44Slide 44:Milestones of Routine (PDUFA) Inspections
DSI is informed of NDA submission DSI and Review Division identifies targets CI sites DSI obtains all pertinent information: protocol/s, CRFs, data listings, 1572, etc. DSI drafts assignment memo to pertinent FDA/ORA Districts ORA personnel schedule & conduct inspection (DSI may participate) DSI is informed of NDA DSI attends filing meeting For paper NDAs, DSI contacts sponsor to obtain additional information about potential sites for clinical investigator inspections, selects sites, and obtains more detailed information about sites to be inspected E-NDAs: DSI may have access to the files, selects sites, and obtains information in electronic format pertaining to the sites of interest. DSI is informed of NDA DSI attends filing meeting For paper NDAs, DSI contacts sponsor to obtain additional information about potential sites for clinical investigator inspections, selects sites, and obtains more detailed information about sites to be inspected E-NDAs: DSI may have access to the files, selects sites, and obtains information in electronic format pertaining to the sites of interest.
45Slide 45:Goals of Inspections: PDUFA
Assess of the following: Clinical Investigator Qualifications Training/Experience/CV review Clinical investigator oversight of study In-depth knowledge of protocol/study plan Selection of competent staff for delegation of responsibilities Clinical Study Center/Site Informed Consent Procedures IRB approval Adherence to study protocol Test article accountability Recordkeeping
Slide 46:The FDA Inspection (Audit) compares Source Document Medical Record Data vs Case Report Forms vs Data Listing Submitted to NDA Verify Source of subjects; Did subjects exist? Did they have the disease under study? Did they meet inclusion/exclusion criteria? IRB Review Obtained? Consent obtained? Adherence to protocol? Verify primary efficacy measurements Adverse events? Safety data: Labs, EKG etc. Drug Accountability? Blinding of data?
What do we look for during the inspection?
CDER BIMO Inspections* FY 2002-2009** *Based on inspection start date **FY09 to Date 556 723 672 690 647 674 667 788 1/5/10Slide 48:What is Generated After an Inspection Is Completed?
ORA may issue a Form FDA-483 at close of inspection to the CI The observations listed on a Form FDA 483 lists inspectional observations Immediately available via FOI ORA Prepares the Establishment Inspection Report (EIR) Prepared by field investigator Includes exhibits supporting all observations including deficiencies Recommends inspection Classification (NAI/VAI/OAI) Submitted to DSI for review DSI Prepares Review of EIR and pertinent exhibits Makes final classification of the inspection (NAI/VAI/OAI) Informs the Review Division (NDA review) via internal report (Clinical Inspection Summary). Includes recommendation on data reliability� DSI Prepares written communication to inspected party (CI): THE LETTER
49Slide 49:For Cause Inspections
50Slide 50:PDUFA vs For-Cause
For-Cause Inspections (Complaints) Based on complaints from any source Allegations that raise concerns regarding data integrity or the rights, welfare, and safety of study subjects have been compromised PDUFA-Related Inspections (NDA) FDA receives a Marketing Application Drug is typically an NME Pivotal studies not conducted under IND (Foreign) Data in support of application is generated only from foreign sites Also may be referred to as �Routine� Inspections
Slide 51:CI Complaint-Related Inspection Assignments* CDER, FY 2001 � 2009**
FY 01 02 03 04 05 06 07 08 09 *Based on Assignment Issued Date **FY09 to Date 1/5/10
52Slide 52:Directed (For Cause) Inspection Criteria
Suspicion of false or fraudulent data Evidence that a sponsor has rejected data from an investigator Evidence of delay in submitting adverse clinical findings Evidence of inadequately monitored clinical investigations Evidence of inadequate or inappropriate informed consent Evidence of delayed or inappropriate IRB approval Evidence that an investigator has a significant financial interest in the product
Slide 53:DSI Complaint Issues
The number of complaints about clinical investigators/clinical trials continues to increase DSI encourages such reporting (new Electronic Complaint Form) Follow-up on complaints is of vital strategic importance Real-time follow-up of real-time issues Public protection; Public confidence
Slide 54:Complaints Received* FY 2001-2009**
01 02 03 04 05 06 07 08 09 *Includes All Branches **FY09 to Date 1/5/10
CDER Clinical Investigator Inspection Assignments* FY 2009** 23% 77% n=492* *Based on assignment issued date **FY09 to Date 1/5/10Slide 56:Clinical Investigator Inspections* All Centers - FY2009
CDER: 458 CBER: 83 CDRH: 163 Total: 704 *Based on Inspection Start Date 1/5/10
57Slide 57:Presentation Outline
Overview of BIMO DSI�s Role & Authority in BIMO Program GCP Guidance vs Regulations The Clinical Investigator Role & Responsibility Selection and Auditing CI Sites PDUFA vs For Cause Post Inspectional Procedures/DSI Actions Consequences Sponsors
58Slide 58:Compliance Classifications
NAI - No Action Indicated Inspected Entity is in compliance VAI - Voluntary Action Indicated Minor deviation(s) from the regulations Voluntary correction is requested OAI - Official Action Indicated Because of serious non-compliance requiring regulatory or administrative action by FDA
Slide 59:Clinical Investigator Inspections Final Classification* FY 2009**
*Based on Letter Issued Date **FY09 to Date Total inspections with final classification = 624 Includes OAI Untitled Letters 1/5/10
Slide 60:CI International Inspections Final Classification* FY 2009**
*Based on Letter Issued Date **FY09 to Date Total inspections with final classification = 120 Includes OAI Untitled Letters 1/5/10
Slide 61:CDER Prevalence of OAI CI Inspections*
Total number of CI inspections: FY09 = 460** FY08 = 373 FY07 = 347 FY06 = 366 FY05 = 403 Total CI inspections =1949 Total OAI cases = 104 Percent OAI = 5.3% *Based on Letter Issued Date **FY09 to Date 1/5/10
62Slide 62:Prevalence of OAI Inspections (DSI) PDUFA vs For Cause
Routine PDUFA inspections FY04 - FY06 = 936 Total OAI cases = 5 Percent OAI = .5% For Cause (Complaints) inspections FY04 - FY06 = 238 Total OAI cases = 25 Percent OAI = 11%
Slide 63:Clinical Investigator Deficiencies CDER Inspections - FY 2009**
48% 53% 37% 41% 29% 12% 27% 6% Foreign n = 120* Domestic n = 401* 11% 5% 2% 3% *Based on Letter Issued Date **FY09 to Date 1/5/10
64Slide 64:Consequences of Non-Compliance: OAI (not all inclusive)
IRBs: Administrative actions; Disqualification Sponsors/CROs/Monitors: Rejection of data; Clinical Holds; Termination of IND; Application Integrity Policy Clinical Investigators: Warning Letters NIDPOE Letters Disqualification
Slide 65:CDER/DSI Clinical Investigator Regulatory Actions
WL = Warning Letter NIDPOE = Notice of Initiation of Disqualification Proceedings and Opportunity to Explain NOOH = Notice of Opportunity for Hearing CA = Consent Agreements (Restricted Agreements) CA = Consent Agreements (Full Disqualification) DQ = Disqualification by Hearing DQ = Disqualification by Commissioner *FY09 to Date 1/5/10
Slide 66:CI Deficiencies - CDER; FY 2009** Final Classification of OAI*
66% 91% 16% (% of total inspections with final classification of OAI; includes OAI:Untitled Letters) 28% 9% N=32 *Based on Letter Issued Date **FY09 to Date 1/5/10
Slide 67:CDER Clinical Investigator OAI Actions (Warning/NIDPOE Letters*) FY 2001-2009**
*Based on Letter Issued **FY09 to Date 1/5/10
Slide 68:CDER OAI Actions (GCP/GLP/BE) Warning/NIDPOE Letters* FY 2001-2009**
*Based on Letter Issued Date **FY09 to Date 1/5/10
69Slide 69:Presentation Outline
Overview of BIMO DSI�s Role & Authority in BIMO Program GCP Guidance vs Regulations The Clinical Investigator Role & Responsibility Selection and Auditing CI Sites PDUFA vs For Cause Post Inspectional Procedures/DSI Actions Consequences Sponsors
70Slide 70:Sponsors & Contract Research Organizations (CROs) Responsibilities
Costs Qualified CIs Regulatory Affairs Protocol Development Protocol Compliance for all Sites Drug Disposition for all Sites Clinical Monitoring AE Reporting Records
71Slide 71:Contract Research Organization (CRO)
A sponsor may transfer any or all obligations to a CRO The transfer of obligations shall be described in writing A CRO that assumes a sponsor obligation is subject to the same regulatory action as a sponsor
Slide 72:Sponsor Inspections*
73 in FY 2009** 43 in FY 2008 25 in FY 2007 *Based on Inspection Start Date **FY09 to Date 1/5/10
Slide 73:Sponsor/CRO Inspections* Final Classification FY 2009**
Total inspections with final classifications = 60 Includes OAI:Untitled Letters *Based on Letter Issued Date **FY09 to Date 1/5/10 1%
Slide 74:CDER BIMO Inspections*(FY 2009**)
CI = 458 BEQ = 128 IRB = 102 GLP = 27 S/M = 73 Total = 788 *Based on Inspection Start Date **FY09 to Date 1/5/10
75Slide 75: DSI Challenges
76Slide 76:GCP Challenges and Initiatives
Increasing requirements outpace resources Increased number of international study sites & inspections. Risk-based approach to inspection site selection Dispersal of responsibility from CI and accountability of study staff and �third parties� in clinical research Linking real-time inspections (CI/Sponsor/IRB)
77Slide 77:GCP Challenges and Initiatives cont
Facilitating electronic data capture while ensuring data integrity Post marketing requirements Oversight of research in vulnerable populations Managing increased number of inspections related to complaints
Slide 78:CDER IRB Inspections* FY 2001 � 2009**
01 02 03 04 05 06 07 08 09 *Based on Inspection Start Date **FY09 to Date 267 293 160 130 123 66 103 72 102 1/5/10
IRB Deficiencies* FY09**: CDER Quorum Records NAI 42% 35% 36% 19% N= 98 Subpart D 17% Written Procedures *Based on Letter Issued Date **FY09 to Date 7% Research Review 1/5/10Slide 80:IRB Classifications* CDER Inspections � FY09**
53% 42% *Based on Letter Issued Date **FY09 to Date N=98 5% 1/5/10
81Slide 81:DSI Homepage: www.fda.gov/cder/offices/dsi Includes links to the Clinical Investigator Inspection List (NEW), Bioresearch Monitoring Information Systems (BMIS) files (NEW), Warning Letters, NIDPOE Letters, Lists of Disqualified or Restricted or Debarred Investigators, Code of Federal Regulations, etc. FDA Homepage: www.fda.gov Includes links to the Federal Register Notices, FDA guidance documents. Compliance Programs: www.fda.gov/ora/compliance_ref/default.htm
Helpful Websites
82Slide 82:Contact Information
DSI Main Telephone: 301-796-3392 DSI Facsimile: 301-847-8748 Joseph P. Salewski: 301-796-3395 Joseph.Salewski@FDA.HHS.GOV