Download
1 / 53
530 likes | 620 Views
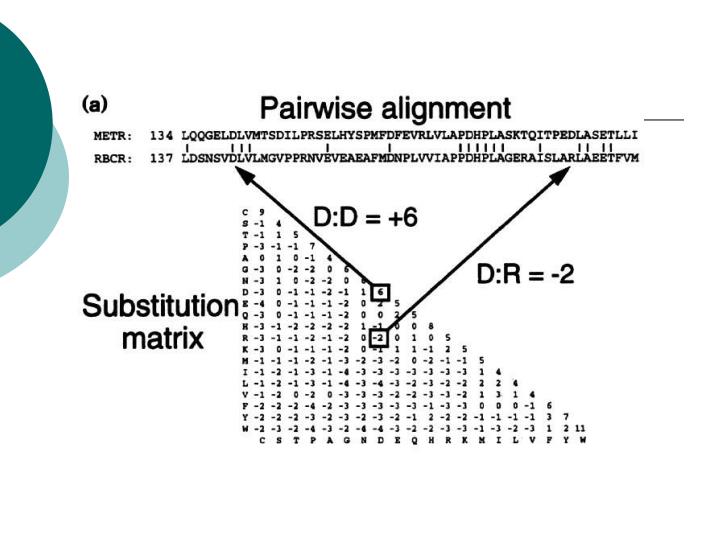
Manual alignment. Difficult for remote homologies Slow Low reproductibility Allows to consider information not included in the sequence. “Equivalent” Aminoácids. Hydrophobic Ala (A), Val (V), Met (M), Leu (L), Ile (I), Phe (F), Trp (W), Tyr (Y) Small Gly (G), Ala (A), Ser (S) Polar

E N D
Manual alignment • Difficult for remote homologies • Slow • Low reproductibility • Allows to consider information not included in the sequence
“Equivalent” Aminoácids • Hydrophobic • Ala (A), Val (V), Met (M), Leu (L), Ile (I), Phe (F), Trp (W), Tyr (Y) • Small • Gly (G), Ala (A), Ser (S) • Polar • Ser (S), Thr (T), Asn (N), Gln (Q), Tyr (Y) • Polar and charged are equivalent on the surface • Charged • Asp (D), Glu (E) / Lys (K), Arg (R) • Hard to replace (special function) • Gly (G), Pro (P), Cys (C), His (H)
BCL2 Human vs BCL2 Mouse >sp|P10417|BCL2_MOUSE Apoptosis regulator Bcl-2 Length=236 Score = 429 bits (1103), Expect = 6e-119, Method: Composition-based stats. Identities = 214/239 (89%), Positives = 217/239 (90%), Gaps = 3/239 (1%) MAHAGRTGYDNREIVMKYIHYKLSQRGYEWDAGDVGAAPPGAAPAPGIFSSQPGHTPHPA 60 MA AGRTGYDNREIVMKYIHYKLSQRGYEWDAGD AAP GAAP PGIFS QP P PA MAQAGRTGYDNREIVMKYIHYKLSQRGYEWDAGDADAAPLGAAPTPGIFSFQPESNPMPA 60 ASRDPVARTSPLQTPAAPGAAAGPALSPVPPVVHLTLRQAGDDFSRRYRRDFAEMSSQLH 120 R+ ARTSPL+ A AGPALSPVPP VHLTLR+AGDDFSRRYRRDFAEMSSQLH VHREMAARTSPLRPLVA---TAGPALSPVPPCVHLTLRRAGDDFSRRYRRDFAEMSSQLH 117 LTPFTARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEY 180 LTPFTARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEY LTPFTARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEY 177 LNRHLHTWIQDNGGWDAFVELYGPSMRPLFDFSWLSLKTLLSLALVGACITLGAYLGHK 239 LNRHLHTWIQDNGGWDAFVELYGPSMRPLFDFSWLSLKTLLSLALVGACITLGAYLGHK LNRHLHTWIQDNGGWDAFVELYGPSMRPLFDFSWLSLKTLLSLALVGACITLGAYLGHK 236 BH4 BH3 BH1 PhosphoSer
BCL-2 vs BCL-X >emb|CAA57886.1| bcl-x [Rattus norvegicus] Length=233 Score = 172 bits (435), Expect = 2e-41, Method: Composition-based stats. Identities = 93/199 (46%), Positives = 116/199 (58%), Gaps = 13/199 (6%) NREIVMKYIHYKLSQRGYEW----DAGDVGAAPPGAAPAPGIFSSQPGHTPHPAASRDPV 66 N+E+V+ ++ YKLSQ+GY W D + P S P + P NQELVVDFLSYKLSQKGYSWSQFSDVEENRTEAPEETEPERETPSAINGNPSWHLADSPA 64 ARTSPLQTPAAPGAAAGPALSPVPPV--VHLTLRQAGDDFSRRYRRDFAEMSSQLHLTPF 124 A G ++ V P+ V LR+AGD+F RYRR F++++SQLH+TP VN-------GATGHSSSLDAREVIPMAAVKQALREAGDEFELRYRRAFSDLTSQLHITPG 117 TARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEYLNRH 184 TA F VV ELFRDGVNWGRIVAFF FGG +CVESV++EM LV IA WM YLN H TAYQSFEQVVNELFRDGVNWGRIVAFFSFGGALCVESVDKEMQVLVSRIASWMATYLNDH 177 LHTWIQDNGGWDAFVELYG 203 L WIQ+NGGWD FV+LYG LEPWIQENGGWDTFVDLYG 196
BCL-2 vs BCL-X >emb|CAA57886.1| bcl-x [Rattus norvegicus] Length=233 Score = 172 bits (435), Expect = 2e-41, Method: Composition-based stats. Identities = 93/199 (46%), Positives = 116/199 (58%), Gaps = 13/199 (6%) NREIVMKYIHYKLSQRGYEW----DAGDVGAAPPGAAPAPGIFSSQPGHTPHPAASRDPV 66 N+E+V+ ++ YKLSQ+GY W D + P S P + P NQELVVDFLSYKLSQKGYSWSQFSDVEENRTEAPEETEPERETPSAINGNPSWHLADSPA 64 ARTSPLQTPAAPGAAAGPALSPVPPV--VHLTLRQAGDDFSRRYRRDFAEMSSQLHLTPF 124 A G ++ V P+ V LR+AGD+F RYRR F++++SQLH+TP VN-------GATGHSSSLDAREVIPMAAVKQALREAGDEFELRYRRAFSDLTSQLHITPG 117 TARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEYLNRH 184 TA F VV ELFRDGVNWGRIVAFF FGG +CVESV++EM LV IA WM YLN H TAYQSFEQVVNELFRDGVNWGRIVAFFSFGGALCVESVDKEMQVLVSRIASWMATYLNDH 177 LHTWIQDNGGWDAFVELYG 203 L WIQ+NGGWD FV+LYG LEPWIQENGGWDTFVDLYG 196 BH4 BH3 BH1 PhosphoSer
Multiple alignment & functional prediction Aim: Predict function from sequence
General strategies • Group sequences in a “family” (BLAST, PFAM) • Recognize sequence fragments (short) related to functional or structural features
Multiple alignment • Global alignment, more than two sequences • Allow to include the importance of the position in the alignment • Allow to define conserved residues • Residues with functional or structural importance • Tree determinants • Correlated mutations
Multiple alignments • Low similarity, only two sequences: AVTTGLNMWTTAKRPGMDDFYTILLPGLMNCIGLFTAIDMHFFGRKPACEEYFTLVVDGLCNCI • Low similarity, multiple sequences: GIFTDIDMHFYVKKPGLDEFFTLVLRTLCMAAALTTGIDMWTTAKRPDMDDYYTIIIPGLMNCIAVTTGLNMWTTAKRPGMDDFYTILLPGLMNCIGVTTGLNMYFTARRPGLDEFYTLVLRTLCMCL GIFTDIDMHFYVKKPGLDEFFTLVLRTLCMAAAVTTGLNMWTTAKRPGMDDFYTILLPGLMNCIGLFTALNMHFFGRKPACEEYFTLVVDGLCNCI
Tree determinants • Define subfamilies • Relevant to philogeny S1 GIFTDIDMHFYVKKPGLDEFFTLVLRTLCMAAS2 ALTTGIDMWTTAKRPDMDDYYTIIIPGLMNCIS3 AVTTGLNMWTTAKRPGMDDFYTILLPGLMNCIS4 GVTTGLNLYFTARRP--DEFYS-VLRTLCMCL S5 GIFTDIDLHFYVKKP--DEFFSLVLRTLCMAAS6 AVTTGLNLWTTAKRP--DDFYSILLPGLMNCIS7 GLFTALNLHFFGRKP--EEYFSLVVDGLCNCI
Correlated mutations • Concerted changes in two or more conserved positions • Reveal positions of structural interaction GIFTDIDMHFYVKKPGL DEFFTLVLRTLCMAAALTTGIDMWTTAKRPDM DDYYTIIIRGLMNCIAVTTGLDMWTTAKRPGM DDFYTILLRGLMNCIGVTTGLDMYFTARRPGL DEFYTLVLKTLCMCL GIFTDIRMHFYVKKPGL DEFFTLVLDTLCMAAAVTTGLRMWTTAKRPGM DDFYTILLDGLMNCIGLFTALRMHFFGRKPAC EEYFTLVVEGLCNCI R-D D-R
Software • ClustalW • Makes global pairwise alignments building “clusters” of similar sequences • Tcoffee • Slower than clustalw but more precise for low similarity • Combines global/local alignments
Profiles • Also known as Position-specific score matrix (PSSM). • Give scores for amino acids or gaps specific to sequence positions • Quantitative approach to include the role of positions
F K L L S H C L L V F K A F G Q T M F Q Y P I V G Q E L L G F P V V K E A I L K F K V L A A V I A D L E F I S E CI I Q F K L L G N V L V C A 0 01 0111 0 1 0 C 0 0 0 0 0 02 0 01 D 0 0 0 0 0 00 0 01 E 0 1 0 0 021 0 0 0 F 5 0 0 1 0 0 0 0 1 0 G 0 0 0 03 0 0 0 01 H 0 0 0 0 01 0 0 0 0 I 0 011 0 0 031 0 K 0 4 0 01 0 0 0 01 L 1 023 0 0 033 0 M 0 0 0 0 0 0 01 0 0 N 0 0 0 0 01 0 0 0 0 P 0 2 0 0 0 0 0 0 0 0 Q 0 0 0 0 02 0 0 02 R 0 0 0 0 0 0 0 0 0 0 S 0 0 0 02 0 0 0 0 0 T 0 0 0 0 0 01 0 0 0 V 0 022 0 02 0 11 W 0 0 0 0 0 0 0 0 0 0 Y 1 0 0 0 0 0 0 0 0 0
Profiles, simplest calculation Frecuency of i at position j Nij/NS Mij = log fi Standard frequency of i. > 0: Position j is rich in aa. i Mij 0: Normal < 0: Position j is poor in the aa. i
Profiles, improvements • Include amino acid frequency according to protein family • Include also classical similarity matrices • Allows equivalent amino acids that do no appear in the available alignment.
F K L L S H C L L V F K A F G Q T M F Q Y P I V G Q E L L G F P V V K E A I L K F K V L A A V I A D L E F I S E CI I Q F K L L G N V L V C A -18 –10 -1 -8 8 -3 3 -10 -2 -8 C -22 -33 -18 -18 -22 -26 22 -24 -19 -7 D -35 0 -32 -33 -7 6 -17 -34 -31 0 E -27 15 -25 -26 -9 23 -9 -24 -23 -1 F 60 -30 12 14 -26 -29 -15 4 12 -29 G -30 -20 -28 -32 28 -14 -23 -33 -27 -5 H -13 -12 -25 -25 -16 14 -22 -22 -23 -10 I 3 -27 21 25 -29 -23 -8 33 19 -23 K -26 25 -25 -27 -6 4 -15 -27 -26 0 L 14 -28 19 27 -27 -20 -9 33 26 -21 M 3 -15 10 14 -17 -10 -9 25 12 -11 N -22 -6 -24 -27 1 8 -15 -24 -24 -4 P -30 24 -26 -28 -14 -10 -22 -24 -26 -18 Q -32 5 -25 -26 -9 24 -16 -17 -23 7 R -18 9 -22 -22 -10 0 -18 -23 -22 -4 S -22 -8 -16 -21 11 2 -1 -24 -19 -4 T -10 -10 -6 -7 -5 -8 2 -10 -7 -11 V 0 -25 22 25 -19 -26 6 19 16 -16 W 9 -25 -18 -19 -25 -27 -34 -20 -17 -28 Y 34 -18 -1 1 -23 -12 -19 0 0 -18
F K L L S H C L L V F K A F G Q T M F Q Y P I V G Q E L L G F P V V K E A I L K F K V L A A V I A D L E F I S E CI I Q F K L L G N V L V C A -18 –10 -1 -8 8 -3 3 -10 -2 -8 C -22 -33 -18 -18 -22 -26 22 -24 -19 -7 D -35 0 -32 -33 -7 6 -17 -34 -31 0 E -27 15 -25 -26 -9 23 -9 -24 -23 -1 F 60 -30 12 14 -26 -29 -15 4 12 -29 G -30 -20 -28 -32 28 -14 -23 -33 -27 -5 H -13 -12 -25 -25 -16 14 -22 -22 -23 -10 I 3 -27 21 25 -29 -23 -8 33 19 -23 K -26 25 -25 -27 -6 4 -15 -27 -26 0 L 14 -28 19 27 -27 -20 -9 33 26 -21 M 3 -15 10 14 -17 -10 -9 25 12 -11 N -22 -6 -24 -27 1 8 -15 -24 -24 -4 P -30 24 -26 -28 -14 -10 -22 -24 -26 -18 Q -32 5 -25 -26 -9 24 -16 -17 -23 7 R -18 9 -22 -22 -10 0 -18 -23 -22 -4 S -22 -8 -16 -21 11 2 -1 -24 -19 -4 T -10 -10 -6 -7 -5 -8 2 -10 -7 -11 V 0 -25 22 25 -19 -26 6 19 16 -16 W 9 -25 -18 -19 -25 -27 -34 -20 -17 -28 Y 34 -18 -1 1 -23 -12 -19 0 0 -18
F K L L S H C L L V F K A F G Q T M F Q Y P I V G Q E L L G F P V V K E A I L K F K V L A A V I A D L E F I S E CI I Q F K L L G N V L V C A -18 –10 -1 -8 8 -3 3 -10 -2 -8 C -22 -33 -18 -18 -22 -26 22 -24 -19 -7 D -35 0 -32 -33 -7 6 -17 -34 -31 0 E -27 15 -25 -26 -9 23 -9 -24 -23 -1 F 60 -30 12 14 -26 -29 -15 4 12 -29 G -30 -20 -28 -32 28 -14 -23 -33 -27 -5 H -13 -12 -25 -25 -16 14 -22 -22 -23 -10 I 3 -27 21 25 -29 -23 -8 33 19 -23 K -26 25 -25 -27 -6 4 -15 -27 -26 0 L 14 -28 19 27 -27 -20 -9 33 26 -21 M 3 -15 10 14 -17 -10 -9 25 12 -11 N -22 -6 -24 -27 1 8 -15 -24 -24 -4 P -30 24 -26 -28 -14 -10 -22 -24 -26 -18 Q -32 5 -25 -26 -9 24 -16 -17 -23 7 R -18 9 -22 -22 -10 0 -18 -23 -22 -4 S -22 -8 -16 -21 11 2 -1 -24 -19 -4 T -10 -10 -6 -7 -5 -8 2 -10 -7 -11 V 0 -25 22 25 -19 -26 6 19 16 -16 W 9 -25 -18 -19 -25 -27 -34 -20 -17 -28 Y 34 -18 -1 1 -23 -12 -19 0 0 -18
Profiles • The use of profiles increases the information available and allow to extract “family” features opposite to individual sequence features
HMM profiles • Statistical models (Hidden Markov Models) to build profiles. • The model is “trained” using multiple alignments to determine evolution probabilities. • They contain a “theoretical machinery” that allows to understand sequence relatioships in a quantitative basis.
The twilight zone • Identity below 25 % • Structural similarity exists but it is difficult to identify it using standard methods
PSI-BLAST • Blast search based on profiles prepared dynamically: • Standard Blast search • Building of a position-specific score matrix (PSSM) from the alignment • New search against the profile • Repeat until self-consistence
PSI-BLAST • PSI-BLAST cannot find what Blast cannot detect • E-value for inclusion/exclusion must be chosen carefully (0.01) • Some times the system goes to non-sense results • Number of iterations is usually kept small
Motifs • Motif: short sequence fragment. Highly conserved, related to some structural or functional feature • Conserved in distant homologues, due to functional or structural restrictions • Help to functional prediction and to detect remote homology
X(12) H C X(3,5) X(2,4) H C C-x(2,4)-C-x(12)-H-x(3,5)-H
Motifs • How to express motifs? • Regular expressions, patterns • Profiles (PSSM) • HMMs (Hidden Markov Models) (PFAM)
Regular expressions ALRDFATHDDF SMTAEATHDSI ECDQAATHEAS A-T-H-[DE]
Regular expressions Never E o D 1 aa [AC]-x-V-x(4)-{ED} A o C 4 aa Sólo V
Regular expressions one or none <A-x-[ST](2)-x(0,1)-V Two S o T N-Terminal
Building regular expressions • From multiple alignments • Manual • Pattern building software • eMotif, PRATT, Teiresias,…
Pattern databases • PROSITE • Known sequence motifs • Active site signatures • Interaction sites • Modification sites (glycosilation, phosphorilation, ...)
[GA]-x(1,2)-[DE]-x-Y-x-[STAP]-x-C-[NKR]-x-[CH]-[LIVMFYWH] G GQ D L Y V P V C R L C Y