Download

1 / 38
890 likes | 2.45k Views
Genetic Polymorphism in Drug Metabolism – CYP450 Isoenzymes. Presented by: Lahari Paladugu ( PharmD 09-10) Presented on: February 7 , 2014. What is Genetic Polymorphism?. (1) The existence together of many forms of DNA sequences at a locus within the population.

E N D
Genetic Polymorphism in Drug Metabolism – CYP450 Isoenzymes Presented by: Lahari Paladugu (PharmD 09-10) Presented on: February 7, 2014
What is Genetic Polymorphism? (1) The existence together of many forms of DNA sequences at a locus within the population. (2) A discontinuous genetic variation that results in different forms or types of individuals among the members of a single species.
Genetic Polymorphism & Drug Metabolism • inter-individual variation of drug effects • Genetic polymorphisms of drug-metabolizing enzymes give rise to distinct subgroups in the population that differ in their ability to perform certain drug biotransformation reactions. • Polymorphisms are generated by mutations in the genes for these enzymes, which cause decreased, increased, or absent enzyme expression or activity by multiple molecular mechanisms.
Drug Metabolism • The metabolism of drugs and other xenobiotics into more hydrophilic metabolites is essential for their elimination from the body, as well as for termination of their biological and pharmacological activity. • Drug metabolism or biotransformation reactions are classified as either phase I functionalization reactions or phase II biosynthetic (conjugation reactions).
Drug Metabolism (cont.) • The enzyme systems involved in the biotransformation of drugs are localized primarily in the liver. • Other organs with significant metabolic capacity include the GI tract, kidneys, and lungs. • These biotransformation reactions are carried out by CYPs (cytochrome CYP450 isoforms) and by a variety of transferases.
Drug Metabolism (cont.) • Pathways of drug metabolism are classified as either: • Phase I reactions: oxidation, reduction, hydrolysis • Phase II reactions: acetylation, glucuronidation, sulfation, methylation • Both types of reactions convert relatively lipid soluble drugs into relatively inactive and more water soluble metabolites, allowing for more efficient systemic elimination.
Polymorphisms • Genetic differences in drug metabolism are the result of genetically based variation in alleles for genes that code for enzymes responsible for the metabolism of drugs. • In polymorphisms, the genes contain abnormal pairs or multiples or abnormal alleles leading to altered enzyme function. • Differences in enzyme activity occur at different rates according to racial group.
Single Nucleotide Polymorphisms (SNPs) • Single changes in one allele of a gene responsible for a variety of metabolic processes including enzymatic metabolism. • The combination of alleles encoding the gene determines the activity and effectiveness of the enzyme. • The overall function of the enzyme is the phenotype of enzyme function. • Phenotype: the observable physical or biochemical characteristics determined by both genetic makeup and environmental influences
Poor metabolizers • two defective alleles (ex: CYP2D6*4/*5 and CYP2D6*4/*4) • Combination of alleles including one resulting in no enzyme (ex: CYP2D6*5 and CYP2D6*4 deletion) • Intermediate metabolizers • Heterozygous – having only one wild type allele and one defective allele • Normal metabolizers • Carry wild type alleles (ex: CYP2D6*1/*3). • Wild type alleles encode genes for normal enzyme function
Extensive metabolizers • Carry one wild type and one amplified gene • ex: CYP2D6*1/*2, CYP2D6*A/*1a, and CYP2D6*1A/*5 • Ultra-rapid metabolizers • Carry two or more copies of amplified gene • ex: CYP2D6*2/*3
Genetic changes may inactivate or reduce enzyme activity leading to increase in the substrate drug. • Genetic duplication may increase enzyme activity resulting in lower levels of substrate drug.
Inhibitors & Inducers • Polymorphisms affect drug interactions by altering the effect of inhibitors and inducers on the enzyme. • results in an exaggerated effect or minimal effect on the substrate • Inhibitor: An enzyme inhibitor is a molecule, which binds to enzymes and decreases their activity. • Inducer: An enzyme inducer is a type of drug that increases the metabolic activity of an enzyme either by binding to the enzyme and activating it, or by increasing the expression of the gene coding for the enzyme.
Extensive Metabolizers - Inhibitors • Extensive metabolizer ----- level of substrate drug is normally low due to rapid metabolism by the enzyme. • An inhibitor to the enzyme will inhibit the extensive metabolism and cause significant elevations in the substrate drug. • Effect of inhibitors is much greater in an EM inc. level of substrate levels
Poor Metabolizers - Inhibitors • In a poor metabolizer, the level of substrate drug remains high because the metabolism of the substrate is much less than normal. • When an inhibitor is added, the additional inhibition of metabolism is not much greater than is already occurring in the PM. • The effect of inhibitor is less in a PM than in normal metabolizers. • The drug interaction might not occur.
Extensive Metabolizers - Inducers • Level of substrate drug is lower than in a normal metabolizer due to rapid metabolism. • The addition of an inducer does not cause a greater difference in the level of substrate because the metabolism is already increased greatly. • The drug interaction might not occur.
Poor Metabolizers - Inducers • Level of substrate drug is higher than expected in normal metabolizer because of the lower metabolism of substrate. • The addition of inducer will cause a signification increase in the metabolism of the substrate much lower level of substrate than expected in a normal metabolizer. • Drug interaction may occur to a greater extent. • Drug interaction may result in substrate levels similar to those of normal metabolizers.
**NOTE** • The effect of inhibitor is great in EMs than in PMs. • The effect of inducer is greater in PMs than in EMs.
Complex Drug Interactions • Can be seen when a substrate is metabolized through more than one enzyme systems where one or more enzymes are affected by polymorphism.
Genetic Polymorphisms in Genes that Can Influence Drug Metabolism – CYP450 Isoforms
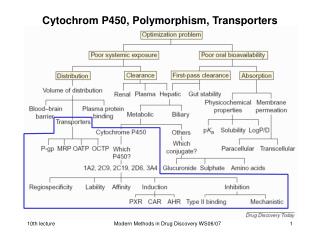
P450 Enzymes in Drug Metabolism • The polymorphic P450 (CYP) enzyme superfamily is the most important system involved in the biotransformation of many endogenous and exogenous substances including drugs, toxins, and carcinogens. • Genotyping for CYP polymorphisms provides important genetic information that help to understand the effects of xenobiotics on human body. • For drug metabolism, the most important polymorphisms are those of the genes coding for CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5, which can result in therapeutic failure or severe adverse reactions.
CYTOCHROME P4502D6 • Most extensively studied polymorphic drug metabolizing enzyme • Debrisoquin --- marked hypotension • Impaired ability to hydroxylate, and therefore, inactivate debrisoquin • 5-10% of white subjects have relative deficiency in ability to oxidize debrisoquin • Also have impaired ability to metabolize the antiarrhythmic and oxytocic drug sparteine • PM lower urinary concentration, higher plasma concentrations • Subjects inherited two copies of a gene or genes that encoded an enzyme with either decreased CYP2D6 activity or no activity at all • Prominent in East African population – frequency as high as 29%
CYTOCHROME P4502C SUBFAMILY • Accounts for approximately 18% of the CYP content in the liver • Catalyzes roughly 20% of the CYP-mediated metabolism of drugs CYP2C19 • Study using mephenytoin as probe drug determined that individuals can be segregated into EMs and PMs. • PM trait is autosomal recessive – present in 3-5% of Caucasians & 12-23% of Asian populations
CYP2C19 (cont.) • Also catalyzes the metabolism of several proton pump inhibitors (i.e. omeprazole), diazepam, thalidomide, and some barbiturates. • Responsible for inactivation or propranolol and metabolic activation of antimalarial drug proquanil.
CYP2C19 & Diazepam • Diazepam is demethylated by CYP2C19 • Plasma diazepam half-life islonger in individuals who are homozygous for the defective CYP2C19*2 allele compared to those who are homozygous for the wild type allele. • Half-life of the desmethyldiazepam metabolite is also longer in CYP2C19 poor metabolizers. • High frequency in Asian population. • Diazepam induced toxicity may occur as a result of slower metabolism – careful dosing in Asian population.
CYP2C9 • Major CYP2C subfamily member in the liver • Primarily responsible for the oxidative metabolism of important compounds – warfarin, phenytoin, tolbutamide, glipizide, losartan, etc. • 6 different polymorphisms – CYP2C9*1, *2, *3, *4, *5, *6 • CYP2C9*1 – wild type allele, CYP2C9*2-*6 – variants • Variants *2 and *3 alleles are common in Caucasians (≈35%) • CYP2C9*2 and *3 alleles associated with significant reduction in the metabolism and clearance of selected CYP2C9 substrates
CYP2C9 & Warfarin • Polymorphisms linked to both toxicity and dosage requirements for optimal anticoagulation with warfarin. • *2 and *3 variants – higher risk of acute bleeding complications than patients with *1 wild type genotype. • Require 15-30% lower maintenance dose of warfarin to achieve target INR • Patients with variant CYP2C9 genotype take a median of 95 days longer to achieve stable dosing compared to wild-type group
Dihydropyrimide Dehydrogenase • Metabolism of antineoplastic agent fluorouracil. • In the 1980s, fatal CNS toxicity developed in several patients after treatment with standard doses fluorouracil. • Patients had inherited deficiency of dihyropyrimidine dehydrogenase. • DPD metabolizes fluorouracil and endogenous pyrimidines. • Severe fluorouracil toxicity occurs when DPD activity < 100 pmol/min/mg protein. • 3% of population carries heterozygous mutations that inactivate DPD and 1% are homozygous for the inactivating mutations. • Heterozygous individuals do not exhibit no phenotype until challenged with fluorouracil.
CYTOCHROME P4503A SUBFAMILY • CYP3A subfamily plays a critical role in the metabolism of more drugs than any other phase I enzyme. • Expressed in liver and small intestine • Contribute to oral absorption, first-pass, and systemic metabolism • Expression is highly inducible – enzyme activity influence by factors such as variable homeostatic control mechanisms, up- or down- regulation by environment factors, and polymorphisms.
CYP3A4 • More than 30 SNPs have been identified for CYP3A4 gene • Unlike other P450s, there is no evidence for deleted or null allele for CYP3A4. • The most common variant in CYP3A4, CYP3A4*1B is an A392G transition in the promoter region referred to as the nifedipine response element. • One study shows that this variant may be associated with a slower clearance of cyclosporine. • This is a rather controversial finding.
CYP3A5 • Polymorphically expressed in adults in about 10-20% in Caucasians, 33% in Japanese, and 55% in African Americans. • The variable CYP3A5*3 is a result of improper mRNA splicing and reduced translation of functional protein. • CYP3A5 is the primary extra-hepatic CYP3A isoform, its polymorphic expression has been implicated in disease risk and the metabolism of endogenous steroids or drug in tissues other than liver. • CYP3A5 has been linked to tacrolimus dose requirements to maintain adequate immunosuppression in solid organ transplant patients.
CYP3A7 • Expressed in fetal liver during development • Hepatic expression is generally down-regulated after birth, but the CYP3A7 protein has been detected in some adults • Increased CYP3A7 expression has been associated with the replacement of 60 nucleotide fragment of the CYP3A7 promoter with the corresponding region form of the CYP3A4 promoter (CYP3A7*1C allele.) • This promoter swap results in increased gene expression of the pregnane X receptor response element.
PXR signaling serves as a central regulator of inducible CYP3A4 expression as well as several other genes involved in drug detoxification. • Polymorphisms in PXR suggest observed variability in CYP3A4 enzymatic activity may be due to, in part, inherited differences in the upstream signaling proteins that control induction of gene expression.
N-ACETYLTRANSFERASE • N-acetylation of isoniazid to acetylisoniazid • Individuals are slow or rapid acetylators • Ethnic variation is seen • Slow acetylation: Japanese (10%), Chinese (20%), Caucasians (60%) • NAT2 protein is the specific protein isoform that acetylates isoniazid. • 27 unique NAT2 alleles identified • NAT2*4 is the wild type allele • NAT2 alleles containing the G191A, T341C, A434C, G590A, and/or G857A missense associated substitutions are associated with slow acetylator phenotype.
References • Shargel, Leon. Chapter 12 –Pharmacogenetics. Applied Biopharmaceutics and Pharmacokinetics, 5th edition. E-book. • Shargel, Leon. Comprehensive Pharmacy Review, 7th Edition. Philadelphia: Lipincott- William & Wilkins, 2010. Print. Pages 430-433. • David B. Troy, Paul Beringer. Remington: The Science and Practice of Pharmacy, 21st Edition. Pages 1230 – 1239. • Brunton, Laurence. Chabner, Bruce. Knollman, Bjorn. Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th edition. Pages 124-130. • http://www.biology-online.org/dictionary/Genetic_polymorphism • http://en.wikipedia.org/wiki/Drug_metabolism • http://www.medscape.com/viewarticle/444804_5 • http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1934960/ • http://dmd.aspetjournals.org/content/29/4/570.full