Download

1 / 144
1.55k likes | 2.17k Views
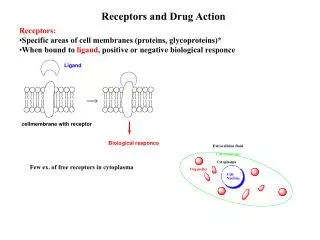
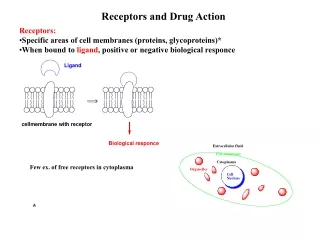
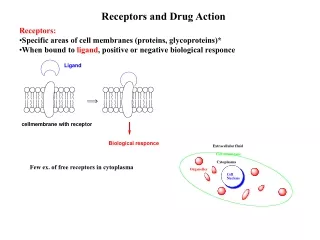
The Organic Chemistry of Drug Design and Drug Action. Chapter 7 Drug Metabolism. Drug Metabolism. Foreign organism – elicits antibody response Low molecular weight xenobiotics – nonspecific enzymes convert them into polar molecules for excretion

E N D
The Organic Chemistry of Drug Design and Drug Action Chapter 7 Drug Metabolism
Drug Metabolism Foreign organism – elicits antibody response Low molecular weight xenobiotics – nonspecific enzymes convert them into polar molecules for excretion Enzymatic biotransformations of drugs – drug metabolism
Principal site of drug metabolism is the liver; also kidneys, lungs, GI tract Pathway of Oral Drugs take via mouth absorbed through small intestine or stomach bloodstream liver (first metabolized) Drug metabolism by liver enzymes – first-pass effect
Avoid first-pass effect by changing the route of administration • sublingual route (under the tongue) bypasses liver - angina (sublingual) • rectal route (suppository or enema) - migraine headaches (rectal) • intravenous (i.v.) injection – rapid response, circulation time of 15 seconds
Avoid first-pass effect by changing the route of administration (cont’d) • intramuscular (i.m.) injection – for large volumes or slow absorption • subcutaneous (s.c.) injection – through loose connective tissue of s.c. layer of skin • pulmonary absorption – gaseous or highly volatile drugs • topical application - asthma (aerosol) Prodrug approaches are discussed in Chapter 8
Drug metabolism is desirable once drug has reached site of action – may produce its effect longer than desired or become toxic. Drug metabolism studies are essential for the safety of drugs. Metabolites must be isolated and shown to be nontoxic.
Synthesis of Radioactive Compounds To increase sensitivity for detection of metabolites, radioactivity is incorporated into the drug candidate. Incorporate a commercial radioactive compound near the end of the synthesis, if possible. Usually the radioactive synthesis is different from that of the unlabeled compound. [14C] preferable to [3H] – 3H has shorter t1/2; isotope effect on C-H cleavage; loss of 3H as 3H2O if C-H cleavage occurs Only a trace amount of radioactivity is used (maybe 1 in 106 molecules); the remainder of the molecules is nonradiolabeled.
If the drug is a natural product, a biosynthetic approach to radioactive incorporation is best Biosynthesis of penicillins Scheme 7.1
If the drug is not a natural product, a chemical synthesis is needed. Synthesis of the Antibacterial Linezolid Scheme 7.2 [14C] acetic anhydride could be used here
The radioactive drug is used in metabolism and bioavailability studies in rats, mice, or guinea pigs, then in dogs and/or monkeys. If >95% of the radioactivity is found in urine and feces, and is nontoxic, it can be administered to humans. Phase I clinical trials on healthy volunteers – radiolabeled drug administered to humans for human metabolism studies.
Advances that Made Metabolism Studies Less Difficult More commercially-available radioactive compounds High performance liquid chromatography (HPLC); new column packings New mass spectrometric methods – tandem mass spectrometry/mass spectrometry; GC/mass spectrometry; *HPLC/electrospray mass spectrometry New nuclear magnetic resonance (NMR) techniques *HPLC/NMR *HPLC/NMR/MS
Principal Steps in Drug Metabolism Studies • Isolation (often, this step can be omitted) – extractions, ion exchange • Separations – HPLC, GC • Identification – mass spectrometry (MS), NMR • Quantification – radioactive labeling, GC, HPLC LC/MS/MS is a rapid method in which a sample is injected into the HPLC, then each peak is run into an electrospray ionization MS for parent ion data, then the parent ion is run into a second MS for fragmentation data.
Pathways for Drug Deactivation and Elimination Rate and pathway of drug metabolism are affected by species, strain, sex, age, hormones, pregnancy, and liver diseases. Drug metabolism is stereoselective, if not stereospecific. Generally, enantiomers act as two different xenobiotics – different metabolites and pharmacokinetics. Sometimes the inactive enantiomer produces toxic metabolites or may inhibit metabolism of active isomer. Metabolism of enantiomers may depend on the route of administration. For example, the antiarrhythmia drug verapamil is 16 times more potent when administered i.v. than orally.
As the lipophilicity increases, metabolism increases; increased lipophilicity leads to better substrate activity with metabolizing enzymes. Figure 7.1
One enantiomer can be metabolized to the other. (Advil) Inactive (R)-isomer is metabolized to active (S)-isomer No need to use a single enantiomer
Drug metabolism reactions – two categories Phase I transformations – introduce or unmask a functional group, e.g., by oxygenation or hydrolysis Phase II transformations – generate highly polar derivatives (called conjugates) for excretion
Phase I TransformationsOxidative Reactions Late 1940s, early 1950s Metabolism of 4-dimethylaminoazobenzene shown to require O2 and a reducing system (NADPH). Called a mixed function oxidase. One atom of O from O2 is incorporated into product; a heme protein is involved. Cytochrome P450 – family of heme enzymes that catalyzes the same reaction on different substrates (isozymes)
Heme-dependent Mixed Function Oxidase Scheme 4.35 Oxidizing agent Reducing agent Activated coenzyme
Reactions Catalyzed by Cytochrome P450 Table 7.1
Drug-Drug Interactions Changes in the pharmacokinetics and metabolism of drugs when multiple drugs are taken together. One drug may inhibit a cytochrome P450, blocking metabolism of another drug. One drug may induce a cytochrome P450, which increases metabolism of other drugs. Active constituent of St. John’s wort (hyperforin, 7.11) activates the pregnane X receptor, which regulates P450 3A4 transcription.
Site of Reactions Catalyzed by P450 • Part of molecule undergoing reaction is determined by: • topography of the active site of the isozyme • degree of steric hindrance of the heme iron-oxo species to the site of reaction • ease of H atom abstraction or electron transfer from the compound
CYP450 activity is variable in the population • CYP450 is found in liver, kidney and lungs. • There are a number of different P450 families, which differ in their substrate and reaction specificity. • 57 human genes for P450 have been indentified. • Individuals also vary in the properties of their P450s. • CYP450 2C9 and 2D6 are responsible for metabolism of about half of all drugs. • Variations in P450s are racially and ethnically distributed. • Pharmacogenomics—how the genetic characteristics of a person influences their response to drugs.
Individual variation in CYP450 2C9 • CYP450 2C9 metabolizes phenytoin, S-warfarin, tolbutamide, losartan, and many nonsteroidal antiinflammatory agents (NSAIDs). • At least 33 alleles of CYP450 2C9 have been discovered. • Most of the mutant alleles of CYP450 2C9 have low or no enzymatic activity.
CYP450 2C9 and tolbutamide metabolism • Tolbutamide is a sulfonylurea antidiabetes drug. • CYP450 2D9 hydroxylates the aromatic methyl to give a much lower activity metabolite. • Individuals with mutant CYP450 2C9 alleles have higher concentrations of tolbutamide in the blood, longer duration of action, and lower blood glucose, so they are more likely to get hypoglycemia.
CYP450 2C9 and warfarin metabolism • Warfarin is an anticoagulant drug which inhibits vitamin K 2,3-epoxide reductase. • (S)-Warfarin is hydroxylated at C-6 and C-7 by CYP450 2C9 to give inactive metabolites. • Mutant alleles of CYP450 2C9 have less activity for hydroxylation of warfarin, so patients with mutant alleles need to have lower doses. • The therapeutic index for warfarin is small even for wild-type patients.
Individual variation in CYP450 2D6 • P450 2D6 metabolizes opiates, antiarrhytmics, tamoxifen and b-blockers, among others. • More than 60 alleles of 2D6 have been discovered. • Some of the alleles of 2D6 have low or no enzymatic activity (PM). • Some of the alleles of 2D6 have intermediate activity (IM). • Some of the alleles of 2D6 have somewhat higher activity (EM). • Some of the alleles of 2D6 have much higher activity than wild-type (UM).
CYP450 2D6 and opiate metabolism • Codeine is O-demethylated to morphine, the active metabolite in analgesia. • PMs can’t convert codeine to morphine, so don’t get analgesia. • UMs convert codeine to morphine very rapidly, so may experience toxicity. • Infants have been poisoned by breast milk from UM mothers taking codeine.
CYP450 2D6 and tamoxifen metabolism • Tamixofen is an antiestrogen used to treat breast cancer. • The metabolite, 4-hydroxytamoxifen, binds about 100-fold more strongly to estrogen receptors. • 2D6 PMs respond poorly to tamoxifen treatment.
Flavin Monooxygenase (another mixed function oxidase) Scheme 4.34 X is N or S Nucleophiles with anionic groups are not substrates
Reactions of Flavin Monooxygenase Table 7.2 Flavin monooxygenase is often more stereoselective than P450
Aromatic Hydroxylation Intermediate in aromatic hydroxylation 1968 National Institutes of Health (NIH) Scheme 7.3 arene oxide isolated
Mechanism for Arene Oxide Formation and Aromatic Hydroxylation (favored over a) Scheme 7.4
Reactions of Arene Oxides Scheme 7.5 toxic effects
Rearrangement of Arene Oxide to Arenol Scheme 7.6 Called the NIH shift
Competing with the NIH Shift Scheme 7.7 deprotonation The more stabilized the carbocation intermediate, the less favored for hydride shift - more deprotonation.
NIH Shift with Groups Other than H Scheme 7.8 p-chloroamphetamine Oxidation of a halogen-substituted aromatic ring is quite rare.
A common approach to slow down or block aromatic hydroxylation is to substitute the phenyl ring with a para-fluorine or para-chlorine (deactivates the ring). The half-life for the anti-inflammatory drug diclofenac (7.21) is 1 h; for fenclofenac (7.22) is >20 h.
NIH Shift of a Nitro Group Scheme 7.9 antiprotozoal
This reaction is electrophilic aromatic substitution Favors electron-donating substituents No aromatic hydroxylation e- withdrawing uricosuric agent
For drugs with 2 aromatic rings, the more e--rich one usually is hydroxylated. hydroxylation here e- withdrawing - antipsychotic
Species Specificity Major hydroxylation metabolites in dogs Maybe a different isozyme pro-R pro-S in humans - antiepilepsy
Mechanism of Epoxide Hydrolase Hydration of Arene Oxide Scheme 7.10 trans-diol anti-attack
Glutathione S-transferase Reaction with Arene Oxide Scheme 7.11
Toxicity from Arene Oxides benzo[a]pyrene Scheme 7.12 Relationship between soot and cancer noted in 1775 - chimney sweeps frequently developed skin cancer alkylation of DNA and RNA
Alkene Epoxidation Scheme 7.13 Also an anticonvulsant anticonvulsant
Toxic Product of Alkene Oxygenation Scheme 7.14 aflatoxin B1 DNA adduct
Oxidation of Carbons Adjacent to sp2 CentersOxygenation next to aromatic sp2 carbon antidepressant
Hydroxylation stereochemistry at C-1 depends on stereochemistry at C-2 in metoprolol. antihypertensive Stereochemistry at C-2 will affect how the molecule binds in P450, which determines which H is closest to the heme iron-oxo species.
Allylic Hydroxylation antiarrhythmic Oxidation gives 7.38 (R = OH)
Oxidation Next to a Carbonyl Group Enantiomer difference in metabolism hydroxylation here for (+)-isomer hydroxylation here for (-)-isomer sedative/hypnotic