Download

1 / 68
680 likes | 870 Views
Protein structure. Predictive methods. Topics Covered. Secondary structure prediction methods 3D fold prediction Ab initio protein structure prediction Homology-based methods of fold recognition Comparative model construction ( aka homology model construction)

E N D
Protein structure Predictive methods
Topics Covered • Secondary structure prediction methods • 3D fold prediction • Ab initio protein structure prediction • Homology-based methods of fold recognition • Comparative model construction (aka homology model construction) • Community evaluation of protein structure prediction • Critical Assessment of protein Fold Prediction (CASP) http://predictioncenter.org/ • EVA (real-time continuous evaluation of protein fold prediction methods) http://cubic.bioc.columbia.edu/eva/ • Astral datasets • Structural Genomics Initiative
Why Protein Structure Prediction? Y 2005 Sequences 2,300,000 Structures 29,000 We know the experimental 3D structure for ~1% of the protein sequences Andras Fiser, Albert Einstein College of Medicine
Desulfovibrio vulgaris Anacystis nidulans Condrus crispus Anabaena 7120 Principles of Protein Structure GFCHIKAYTRLIMVG… evolution folding Fold Recognition Comparative Modeling Ab initio prediction Andras Fiser, Albert Einstein College of Medicine
Ab initio prediction Comparative Modeling Applicable to any sequence Not very accurate (>4 Ang RMSD), Attempted for proteins of <100 residues Accuracy and applicability are limited by our understanding of the protein folding problem Applicableto those sequences only that share recognizable similarity to a template structure Fairly accurate ( <3 Ang RMSD), typically comparable to a low resolution X-ray experiment. Not limited by size Accuracy and applicability are rather limited by the number of known folds Protein structure modeling Andras Fiser, Albert Einstein College of Medicine
Structural Genomics Definition: The aim of structural genomics is to put every protein sequence within a “modeling distance” of a known protein structure. Size of the problem: There are a few thousand domain fold families. There are ~20,000 sequence families (30% sequence id). Solution: Determine protein structures for as many different families as possible. Model the rest of the family members using comparative modeling Andras Fiser, Albert Einstein College of Medicine
Structural Genomics Characterize most protein sequences (red) based on related known structures (green). The number of “families” is much smaller than the number of proteins Andras Fiser, Albert Einstein College of Medicine
The utility of a comparative model depends on its accuracy Accuracy is closely linked to sequence similarity David Baker and Andrej Sali, Protein Structure Prediction and Structural Genomics, Science 2001
Ca RMSD Å (% EQV) 2 (50) 1 (80) 0 (100) Anacystis nidulans Anabaena7120 COMPARATIVE MODELING Condruscrispus Desulfovibrio vulgaris Clostridium mp. 20 50 100 % SEQUENCE IDENTITY Comparative Protein Structure Modeling Flavodoxin family KIGIFFSTSTGNTTEVA… Andras Fiser, Albert Einstein College of Medicine
TARGET TEMPLATE ASILPKRLFGNCEQTSDEGLKIERTPLVPHISAQNVCLKIDDVPERLIPERASFQWMNDK ASILPKRLFGNCEQTSDEGLKIERTPLVPHISAQNVCLKIDDVPERLIPE MSVIPKRLYGNCEQTSEEAIRIEDSPIV---TADLVCLKIDEIPERLVGE Steps in Comparative Protein Structure Modeling START Template Search Target – Template Alignment Model Building Model Evaluation No OK? Yes END Andras Fiser, Albert Einstein College of Medicine
Incorrect template Misalignment Distortion in correctly aligned regions Region without a template Side chain packing Typical Errors in Comparative Models MODEL X-RAY TEMPLATE Andras Fiser, Albert Einstein College of Medicine
Template identification START Template Search • Fast but less sensitive: e.g. BLAST • Better: Intermediate sequence search • Even better: Profile/HMM and iterative search methods (e.g. PSI-BLAST) • Searching against libraries of HMMs and profiles for solved structures • Profile-profile alignment (e.g., Hhalign, PHYRE) • Including 2ary structure prediction • Structure-based threading Target – Template Alignment Model Building Model Evaluation No OK? Yes END
Target-template alignment START Template Search • Note that the methods for identifying candidate templates normally produce an alignment • but these alignments are unlikely to be optimal • The alignment method used must be tuned to the level of evolutionary divergence between the target and template • Manual refinement/editing of the alignment is often used to improve the comparative model Target – Template Alignment Model Building Model Evaluation No OK? Yes END
Constructing a comparative model START Template Search • Rigid Body Assembly (COMPOSER) • Segment Matching (SEGMOD, 3DPSSM) • Satisfaction of Spatial Restraints (MODELLER) • Integrated (NEST) loop modeling, side chain modeling Target – Template Alignment Model Building Model Evaluation No OK? Yes END Andras Fiser, Albert Einstein College of Medicine
Comparative model evaluation START • Stereochemistry (PROCHECK, WHATCHECK) • Environment (Profiles3D, Verify3d) • Statistical potentials based methods (PROSAII) • Is the model reliable? • A model is reliable when it is based on a • correct template and on an approximately • correct alignment. Template Search Target – Template Alignment Model Building Model Evaluation No OK? Yes END Andras Fiser, Albert Einstein College of Medicine
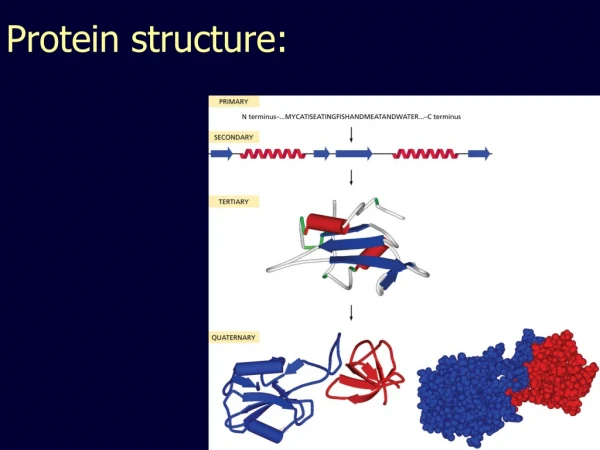
Hierarchical descriptions of proteins(follows the folding process) • Primary structure: the amino acid sequence • Secondary structure: “regular local structure of linear segments of polypeptide chains” (Creighton) • Helix (~35% of residues): subtypes: , and 310 • Beta sheet (~25% of residues) • Both types predicted by Linus Pauling (Corey and Pauling, 1953; helix first described by Pauling in 1951) • Other less common structures: • Beta turns • 3/10 helices • Ω loops • Remaining unclassifiable regions sometimes termed “random coil” or “unstructured regions” • Tertiary structure: “Overall topology of the folded polypeptide chain” (Creighton) • Mediated by hydrophobic interactions between distant parts of protein • Quaternary structure: “Aggregation of the separate polypeptide chains of a protein” (Creighton) Baxevanis & Ouellette (Ch. 9, p.224, Wishart)
Information required for folding is (mostly) contained in the primary sequence • Early on, proteins were shown to fold into their native structures in isolation • This led to the belief that structure is determined by sequence alone (Anfinsen, 1973) • Over the last decade, a significant number of proteins have been shown to not fold properly in the test tube (e.g., requiring the assistance of chaperonins) • Nevertheless, the native 3D structure is assumed to be in some energetic minimum • This led to the development of ab initio folding methods Baxevanis & Ouellette (Ch. 9, Wishart)
Folding pathways • Evidence that local structure segments form first, and then pack against each other to form 3D fold • Exploited in protein fold prediction, Rosetta method • Simons, Bonneau, Ruczinski & Baker (1999). Ab initio Protein Structure Prediction of CASP III Targets Using ROSETTA. Proteins • Semi-stable structural intermediates on folding pathway to lowest-energy conformation • Prof. Susan Marqusee, Berkeley Baxevanis & Ouellette (Ch. 9, Wishart)
Why is secondary structure prediction important? • Secondary structure diverges less rapidly than primary sequence • Knowledge or prediction of 2ary structure improves detection and alignment of remote homologs • 3d-pssm, PHYRE, SAM T02 (fold prediction servers) Baxevanis & Ouellette (Ch. 9, Wishart)
Basic types of secondary structure • Helices ( and others) • is most common; 3.6 residues/turn • Side chains project outward • Structure is stabilized between hydrogen bonds between the carbonyl (CO) group of one amino acid and the amino (NH) group of the amino acid that is 4 positions C-terminal to it • -Strands (two or more strands interact to form a -sheet) • Other (sometimes called loop, coil, or non-regular) • Most secondary structure prediction methods classify residues to one of three states Baxevanis & Ouellette (Ch. 9, Wishart)
Focusing on single residues • Early structure prediction methods focused on the structural characteristics of individual residues • This enabled the larger problem to be decomposed into smaller easier-to-solve problems (enabling the combination of solutions to sub-problems to form a global solution) • This also enabled methods to focus on detecting transmembrane regions, solvent-accessible residues, and other important features of molecules Baxevanis & Ouellette (Ch. 9, Wishart)
Secondary structure prediction accuracy is boosted by using homologs • Labeling residues in a sequence as -helix, -sheet or turn/coil (3-state prediction). • Accuracy of prediction enhanced by ~6% when multiple sequence alignments are used vs the use of a single sequence (Cuff & Barton, 1999) • Best methods for 2ary structure prediction -- PSIPRED (Jones 1999) and JNET (Cuff & Barton, unpublished) • Make use of homologs obtained using PSI-BLAST • Have ~>76% accuracy for 3-state prediction • Provide confidence values for each position Baxevanis & Ouellette (Ch. 12, Barton)
Amino acid patterns indicative of -strand structures • Short runs of conserved hydrophobic • Buried -strand • An i, i+2, i+4 pattern of conserved hydrophobic residues suggests a surface -strand. • Conserved residues sharing the same physicochemical properties are likely to form one face of a strand. Baxevanis & Ouellette (Ch. 12, Barton)
Amino acid patterns indicative of -helical structures • Conservation patterns of i, i+3, i+4, i+7 and variations (e.g., i, i+4, i+7) suggests an alpha helix • Amphiphilic/amphipathic conservation patterns (alternating hydrophobic and polar residues) following an i, i+3, i+4, i+7 pattern (and variations, e.g., i, i+4, i+7) are likely to represent surface helices Baxevanis & Ouellette (Ch. 12, Barton)
Identifying loop regions • Insertions and deletions are not well tolerated in the hydrophobic core. • Regions of an MSA that include many gap characters are likely to indicate surface loops. • Glycine and proline residues can be found in any secondary structure. • However, conserved glycine/proline residues are strongly suggestive of loops. Baxevanis & Ouellette (Ch. 12, Barton)
Amino acid preferences for different secondary structures(and identifying loops/turns) http://www.chembio.uoguelph.ca/educmat/phy456/456lec01.htm
Early schemes used observed preferences • Various schemes give the amino acids numerical weights or rankings for their preferences, and several computer programs can predict the secondary structure from the given sequence. • Preferences are weak, but provide some signal • The simplest such scheme of Chou and Fasman, Ann. Rev Biochem. (1978), examined the statistical distribution of amino acids in alpha helix, beta sheet and turns or loops, using a set of known protein structures from the protein databank. • A novel sequence can then be scanned, and the tendency of each portion of the sequence to form secondary structure is assessed. http://www.chembio.uoguelph.ca/educmat/phy456/456lec01.htm
Improving secondary structure prediction • Peer pressure (pressure from the neighbors): A minimum of 4 amino acids out of 6 should show alpha preference, or 3 out of 5 beta preference, or clusters of 2-3 breakers in a sequence of 4 are needed to set the secondary structure in any region, and individual misfits adopt the secondary structure of their neighbours. • Learning secondary structure preferences from expanded data sets: More recent prediction schemes take advantage of larger data sets to examine amino acid preference for different regions in a helix or different positions in a tight turn. • Up-weighting conserved residues: In addition, sequences of homologous proteins may be compared. The rationale is that highly conserved amino acids contribute more to the three dimensional structure than unconserved, and different weightings can be introduced to the statistical analysis. • Improved accuracy: The accuracy of prediction has risen from about 55% using the simple Chou-Fasman method, where the tendency is to overpredict, to almost 80% using current methods. http://www.chembio.uoguelph.ca/educmat/phy456/456lec01.htm
Amino acid propensities for different structural environments • Propensities are weak but contribute to prediction accuracy • E.g., Glu (E) occurs in alpha helices only 59% more frequently than random • Helical propensities • Partial charge of helix dipole favors • Acidic Asp (D) and Glu (E) residues at N-terminus of helices • Basic Lys (K), Arg (R ) and His (H) residues at C-terminus • Pro (P) residues are more common at the N-terminal first turn of helix • Asn (N), Asp (D), Ser (S) and Thr (T) residues often occur at first turn of helix (side chain hydrogen bonding to backbone of third residue) Creighton, Proteins
The new generation of secondary structure prediction • Based on machine learning concepts • Training set: learn implicit rules, principles and model parameters from labelled data (sequences whose secondary structures are known for each position) • Test set: sequences of unknown structure • Used machine learning method called artificial neural networks(designed to simulate biological neural networks in the brain) • PHDsec (Rost et al 1994, Rost et al 1996) Baxevanis & Ouellette Ch 8 (Ofran and Rost)
Key to success in machine learning algorithms • “The success of machine learning algorithms depends on the careful choice of the biologically based features used for training… and a sufficiently large and accurate training set” • To enhance prediction accuracy on novel data, training data diversity is also critical • Exploit knowledge that local environment is important: to predict 2ary structure of residue ‘i’, consider all residues in a window around i:i-n, … i, … i+n. Baxevanis & Ouellette Ch 8 (Ofran and Rost)
PHDsec • Employs homology detection and a feed-forward artificial neural network • Step 1: homolog search and MSA construction • Step 2: label each position with conservation signal (across MSA) and observed substitutions • Step 3: submit representative annotated “sequence” to a system of neural networks. • Output is a prediction of the most likely secondary structure at each position, with the estimated confidence in that prediction Baxevanis & Ouellette Ch 8 (Ofran and Rost)
Assessing performance evaluations • “Overall, the correct evaluation of performance for prediction methods is an art in itself; only a handful of methods turned out over time to not have been overestimated by their developers.” • Evaluation must be performed on a standard dataset • Training and test data should be rigorously kept separate • Standard deviations of estimates should be provided Baxevanis & Ouellette Ch 8 (Ofran and Rost)
Other problems with comparing different methods • Performance reported in literature can take different forms • Accuracy and coverage • Positive (or negative) predictive power • Sensitivity and specificity • Machine learning terms (e.g., Matthews coefficients) • Wilcoxon paired score signed rank tests • Or might be based on different criteria for success • per residue • per secondary structure element • per protein • Others measure performance only in cases where a prediction has high confidence (with a likelihood of a lower FP rate) Baxevanis & Ouellette Ch 8 (Ofran and Rost)
How do the methods compare? • Best methods now reach 76% accuracy at 3-state prediction (helix, strand, random coil) • Rost 2001 • See EVA website for detailed comparisons • Metaservers: • Consensus approaches combining weighted predictions from different servers • These almost always outperform individual methods • Shown in both CASP and EVA Baxevanis & Ouellette Ch 8 (Ofran and Rost)
Caveats • Even when an experimental structure is available, it is sometimes unclear where one secondary structure element ends and another begins • Low-confidence predictions (and regions of disagreement across servers) can correspond to structurally ambiguous regions • Real-life example: Prion protein (involved in bovine spongiform encephalopathy, Creutzfeld-Jakob disease, etc). • Region assumed to be responsible for aggregation believed to flip from experimentally determined helical structure to (predicted) strand in diseased individuals • All the best secondary structure prediction methods predict this region to be beta (“incorrect”) Baxevanis & Ouellette Ch 8 (Ofran and Rost)
Secondary structure prediction programs • PSI-PRED (David Jones; makes use of distant homologs detected using PSI-BLAST - most popular) • JNET (Cuff & Barton) • PHD (Rost & Sander) Baxevanis & Ouellette Ch 8 (Ofran and Rost)
Consensus and jury approaches produce best results • Primary conclusion of CASP experiments is that structure prediction meta-servers (which combine results from several independent prediction methods) have the highest accuracy • This kind of consensus approach can be applied to both the template selection and the pairwise alignment between the target and template • We have also shown in class that a consensus approach can be applied to predicted structures for numerous homologs in a family related to the target
Basic premise: The function and structure of a protein are encoded in its primary sequence The amino acid sequence determines • a protein’s 3D structure, • subcellular localization, • intermolecular interactions, • biochemical physiological tasks, and • (eventually) how and when it will be broken down into its component building blocks. • Paraphrased from class text (Ofran and Rost), p 198
How many unique protein folds are there? • Many structural biologists believe that all protein domains will eventually be classified into only 1,000 different fold classes (Koonin et al 2002) • Number of unique SCOP folds already close to 1,000 • Structural Genomics Initiative is designed to populate that fold space • However: even with attempts to solve novel structures, many new structures are clearly members of existing structural classes Baxevanis & Ouellette (Ch. 9, Wishart)
3D structure classification schemes • Structure classification databases: • SCOP (http://scop.berkeley.edu/ • CATH (http://www.cathdb.info/) • Three main classes for folds • All alpha (>50% helix; <10% beta sheet) • All beta (>30% beta sheet; <5% helix) • Mixed or alpha/beta (everything else) Baxevanis & Ouellette (Ch. 9, Wishart)
3D structure prediction • Decompose into two subtasks • Fold recognition“Protein X is related by evolution to structure Y” • Assumed evolutionary relationship is used to infer a similarity in 3D fold (but no comparative model construction) • Can be achieved by pairwise sequence comparison, scoring a sequence against a library of profiles or HMMs, and by other methods • Newer “threading” methods can enable correct fold recognition in the Twilight Zone • Comparative model construction • May be restricted to higher sequence identity (e.g., above 30%) due to the likelihood of serious alignment error below this range. • Some servers do both • 3d-pssm/PHYRE, Superfamily, etc. Baxevanis & Ouellette Ch 8 (Ofran and Rost)
Threading • Limited to generating approximate models or suggesting approximate folds • >5 Angstroms for 3D threading • >3Angstroms for 2D threading • Name based on “threading” a tube (called a snake) through a plumbing system. • Each unique threading of a sequence through the 3D model can be evaluated using empirically derived energy function or measure of packing efficiency • Sequences can be scored based on how well they fit the model (i.e., the best score achievable) Baxevanis & Ouellette Ch 9 (Wishart)
Three-dimensional threading • First described by Novotny et al (1984) • Rediscovered in early 1990s • Jones et al 2992; Sippl & Weitckus 1992; Bryant & Lawrence 1993 • Based largely on heuristic contact potentials (interactions between pairs of residues) • 3D coordinates of theoretical structure (based on threading of sequence through PDB structure model) used to evaluate predicted contacts and derive a fitness score based on a pseudoenergy function • Powerful for predicting 3D structure of unknown proteins, and for evaluating structure of known proteins • Limitations found in this method: • interactions are not always conserved between distant homologs • Computational complexity (very slow) • Modest accuracy (early methods ignored amino acid information; model accuracy >5Angstroms) Baxevanis & Ouellette Ch 9 (Wishart)