Download

1 / 31
320 likes | 482 Views
Clustering Large Data Sets in Gene expression analysis Daniel Weaver. Overview. What is “Gene Expression”? Scientific questions and clustering techniques . “The Central Dogma”. The arrows represent the transfer or flow of information.

E N D
Clustering Large Data Sets in Gene expression analysisDaniel Weaver
Overview • What is “Gene Expression”? • Scientific questions and clustering techniques
“The Central Dogma” • The arrows represent the transfer or flow of information. • DNA and RNA store information in a base-4 code (the four nucleotides). • Proteins store information in a base-20 code (the 20 amino acids). Transcription Translation DNA RNA Protein
What’s in a name? • DNARNA = “Transcription” • because the information is exactly copied (or “transcribed”) from one base-4 system (DNA) to an equivalent base-4 system (RNA). Think of a monk transcribing a scroll. • RNAProtein = “Translation” • because the information is converted from a base-4 system (RNA) to a base-20 system (protein). Think of a monk translating a scroll into a new language.
What is a “gene”? • “A gene is a segment of DNA that contains all the information necessary to code for some function.” • A gene is also the unit of information that is transferred through Transcription and Translation.
Switching genes on (or off) • Purpose: to correctly control the amount of active functional (protein) product present in the cell or organism. Promoter Enhancer Figure taken, with permission from Alberts et al., Molecular Biology of the Cell
Presence vs. expression • All cells have the same set of genes. • Different cell types express different subsets of their genes. • Constitutive genes are expressed in most cell types. • Cell-type specific genes are expressed in only a few cell types. A B C A B C
Gene expression responds to the environment • Changes to the cell’s internal or external environment can lead to changes in gene expression. • Most human diseases manifest through a mis-regulation of gene expression A B C A B C
Example - raw microarray data = more abundant in cell type A = more abundant in cell type B = equally abundant in both cell types
log (ratioi) [log2(ratioi)]½ Interpreting raw data • Most gene expression detection data sets are expressed as a ratio of Red:Green (experiment:control) signal. • Frequently use a normalized log(red:green) ratio: for gene X Xi = Such that the Euclidean length of X is 1. • Interpreted raw data are tabulated in a Entity-by-Entity table, Genes-by-Experiments.
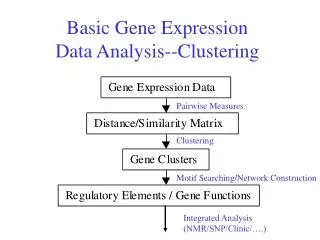
Gene-by-Experiment table • Gene expression analysis is a variant of classic data mining – looking for informative patterns in the rows and columns of this type of table.
Data volumes • ~120,000 genes in the human genome. • Expression detection techniques can take from 1-50 measurement simultaneously on each gene. • Many, diverse Gene and Experiment attributes • In 3-5 years, 105+ data sets will be available for analysis • Data volumes ranging from 10’s of Gb to a few Tb
Analyzing Gene expression data • What genes are (or are not) expressed? • In different cells • Under different external conditions • In different disease states • How much does their expression change? • Does the change in expression correlate with other observed parameters? • Handled with descriptive statistics
Clustering and Classifying gene expression • Scientific questions to be answered • Clustering techniques that are being applied • Lots of room and need for novel statistical and computational analyses
Clustering Gene expression data • Functionally classify novel genes • Identify co-regulated gene groups • Identify diagnostic gene expression patterns
Functionally Classifying Genes • Problem: • Genome sequencing projects identify many, previously unstudied genes. • Can one use the genes’ expression patterns to cluster genes that have similar function?
Inputs and outputs • Inputs • A set of genes whose functional classification is know. • A set of genes whose functional classification is unknown. • Gene expression data sets for all the genes. • Desired Output • A “best fit” functional classification for each of the novel genes.
Examples • Brown et al. (2000) PNAS 97(1), 262-267. • Input: • Log normalized data from 79 experiments on 2,467 genes • Trained on 2/3 of the genes, tested on remaining 3rd. • Classifiers tried include: Support Vector Machines and four machine learning algorithms (Parzen, FLD, C4.5, MOC1 ) • SVM’s performed the best and using the kernel: K(X,Y) = (XY+1)d (d=1,2,or 3) • This kernel transforms the data into higher dimensional space where it is easier to identify a separating hyperplane • Sensitivity = ~0.6
Co-regulated genes • Problem: • Biological processes typically involve genes of many functional categories. • Knowledge of what genes act coordinately can help direct drug development Expression Group 1 Expression Group 2 Expression Group 3
Inputs and Outputs • Inputs • Gene expression data for all genes of interest • (Information about the experimental conditions in which the gene expression data sets were collected) • Desired Outputs • Ordering of the input genes into sets of genes with related expression patterns
Examples • Eisen et al. (1998) PNAS 95: 14863-14868 • Input: • Log normalized data from 12 experiments on 2,467 genes • Performed pair-wise average linkage cluster analysis, using a modified Pearson correlation coefficient metric • Gene that cluster together are displayed in a dendrogram wherein the branch lengths correlate to the degree of similarity
Examples • Tavazoie et al. (1999) Nature Genetics 22:281-285. • Inputs: • “Variance-normalized” data from 15 experiments on 6,220 genes. Variance normalization is Xij = (Xij – Xi)/stdev(Xi) for gene i in experiment j. • Used Euclidean distance as the metric and performed k-means clustering, programmed to find 10, 30, and 60 centroids. • Gene clusters were shown to contain functionally related genes as expected.
Diagnostic expression patterns • Problem: • Many diseases cannot be reliably distinguished through traditional techniques (microscopy, pathology, etc.) • Given gene expression data from diseased tissue, is there a set of genes that correctly distinguishes the diseases (as judged by other criteria).
Inputs and Outputs • Inputs • Gene expression data for all genes (available) • Information about the patients afflicted with the complex disease of interest. • Desired output • The minimal set of genes that accurately partitions the disease, i.e. the minimal diagnostic gene expression pattern.
Examples • Alizadeh et al. (2000) Nature 403: 503-511. • Input: • Log normalized data from 96 experiments on 4,026 genes (out of 17,856 measured). • The 96 experiments were performed on cancer biopsies from patients with Diffuse Large B-cell Lymphoma (DLBCL). • Pair-wise average linkage cluster analysis, using a modified Pearson correlation coefficient metric (Eisen et al., 1998). • Two previously unknown DLBCL sub-types distinguished by small gene clusters (~40 genes and ~70 genes) • Subtypes correspond to prognosis: • “GC B-like” 76% survivorship • “Activated B-like” 16% survivorship (Overhead)
Summary • Current techniques include supervised and unsupervised classification • Three main scientific questions: • Functionally classifying genes • Identifying co-regulated sets of genes • Identifying diagnostic expression “fingerprints” • Data sets are relatively small now, but growing rapidly. • Classification draws from the expression data and from other domain knowledge. • Lots of room and need for novel statistical and computational analyses
Further Reading Clustering Gene Expression Data • Alizadeh, et al. (2000) Nature 403: 503-511. • Alon, et al. (1999) PNAS 96: 6745-6750. • Butte and Kohane. (2000) Proceedings of Pacific Sym. Biocomputing. • Brown, et al. (2000) PNAS 97: 262-267. • Eisen, et al. (1998) PNAS 95: 14863-14868. • Iyer, et al. (1999) Science 283: 83-87. • Raychaudhuri, et al. (2000) Proceedings of Pacific Sym. Biocomputing. • Roberts, et al. (2000) Science 287: 873-880. • Ross et al. (2000) Nature Genetics 24:227-235. • Scherf, et al. (2000) Nature Genetics 24: 236-244. • Spellman, et al. (1998) Mol Biol Cell 9: 3273-3297. • Tamayo, et al. (1999) PNAS 96: 2907-2912. • Tavazoie, et al. (1999) Nature Gen 22: 281-285. • Zhu and Zhang. (2000) Proceedings of Pacific Sym. Biocomputing.
Further Reading Other related gene expression papers: • Holstege, et al. (1998) Cell 95:717-728. • DeRisi et al. (1996) Nature Genetics 14:457-460. • Schena et al. (1995) Science 270:467-470. • DeRisi et al. (1997) Science 278:680-686. • Hilsenbeck et al. (1999) J. Natl. Cancer Inst. 91:453-459.
Expression Data sets • European Bioinformatics Institute (EBI) (links to refs. 4,5,6,11) • Main microarray page • http://www.ebi.ac.uk/microarray/ • Microarray public data set page (this is a great portal site from which you can browse to many of the published data sets) • http://industry.ebi.ac.uk/~brazma/Data-mining/microarray.html • National Human Genome Research Institute (NHGRI) • Main page • http://www.nhgri.nih.gov/DIR/LCG/15K/HTML/ • Data set down load page • ftp://kronos.nhgri.nih.gov/pub/outgoing/olga/old/ • National Cancer Institute (NCI) (ref. 9 & 10) • Main page • http://discover.nci.nih.gov/ • Data set down load page • http://discover.nci.nih.gov/nature2000/ • Lymphoma data set (ref. 1) • Main page • http://llmpp.nih.gov/ • Data set download page • http://llmpp.nih.gov/lymphoma/