Download

1 / 38
390 likes | 635 Views
Assignment and prediction. Protein Secondary Structures. Pernille Andersen 23.04.2007. Outline. What is protein secondary structure How can it be used? Different prediction methods Alignment to homologues Propensity methods Neural networks Evaluation of prediction methods

E N D
Assignment and prediction Protein Secondary Structures Pernille Andersen 23.04.2007
Outline • What is protein secondary structure • How can it be used? • Different prediction methods • Alignment to homologues • Propensity methods • Neural networks • Evaluation of prediction methods • Links to prediction servers
Helix Bend Turn Secondary Structure Elements ß-strand
Use of secondary structure • Classification of protein structures • Definition of loops (active sites) • Use in fold recognition methods • Improvements of alignments • Definition of domain boundaries • Input for a number of alterntive bioinformatics tools
Classification of secondary structure • Defining features • Dihedral angles • Hydrogen bonds • Geometry • Assigned manually by crystallographers or • Automatic • DSSP (Kabsch & Sander,1983) • STRIDE (Frishman & Argos, 1995) • DSSPcont (Andersen et al., 2002)
From http://www.imb-jena.de phi - dihedral angle of the N-Calpha bond psi - dihedral angle of the Calpha-C bond omega - dihedral angle of the C-N (peptide) bond Dihedral Angles
Helices phi(deg) psi(deg) H-bond pattern ----------------------------------------------------------- alpha-helix -57.8 -47.0 i+4 pi-helix -57.1 -69.7 i+5 310 helix -74.0 -4.0 i+3 (omega = 180 deg ) From http://www.imb-jena.de
phi(deg) psi(deg) omega (deg) ------------------------------------------------------------------ beta strand -120 120 180 From http://broccoli.mfn.ki.se/pps_course_96/ Beta Strands Antiparallel Parallel
Helix Bend Turn Secondary Structure Elements ß-strand
* H = alpha helix * G = 310 - helix * I = 5 helix (pi helix) * E = extended strand, participates in beta ladder * B = residue in isolated beta-bridge * T = hydrogen bonded turn * S = bend * C = coil Secondary Structure Type Descriptions
Automatic assignment programs • DSSP ( http://www.cmbi.kun.nl/gv/dssp/ ) • STRIDE (http://bioweb.pasteur.fr/seqanal/interfaces/stride.html) • DSSPcont ( http://cubic.bioc.columbia.edu/services/DSSPcont/ ) • The protein data bank visualizes DSSP assignments on structures in the data base (go to sequence details tab) # RESIDUE AA STRUCTURE BP1 BP2 ACC N-H-->O O-->H-N N-H-->O O-->H-N TCO KAPPA ALPHA PHI PSI X-CA Y-CA Z-CA 1 4 A E 0 0 205 0, 0.0 2,-0.3 0, 0.0 0, 0.0 0.000 360.0 360.0 360.0 113.5 5.7 42.2 25.1 2 5 A H - 0 0 127 2, 0.0 2,-0.4 21, 0.0 21, 0.0 -0.987 360.0-152.8-149.1 154.0 9.4 41.3 24.7 3 6 A V - 0 0 66 -2,-0.3 21,-2.6 2, 0.0 2,-0.5 -0.995 4.6-170.2-134.3 126.3 11.5 38.4 23.5 4 7 A I E -A 23 0A 106 -2,-0.4 2,-0.4 19,-0.2 19,-0.2 -0.976 13.9-170.8-114.8 126.6 15.0 37.6 24.5 5 8 A I E -A 22 0A 74 17,-2.8 17,-2.8 -2,-0.5 2,-0.9 -0.972 20.8-158.4-125.4 129.1 16.6 34.9 22.4 6 9 A Q E -A 21 0A 86 -2,-0.4 2,-0.4 15,-0.2 15,-0.2 -0.910 29.5-170.4 -98.9 106.4 19.9 33.0 23.0 7 10 A A E +A 20 0A 18 13,-2.5 13,-2.5 -2,-0.9 2,-0.3 -0.852 11.5 172.8-108.1 141.7 20.7 31.8 19.5 8 11 A E E +A 19 0A 63 -2,-0.4 2,-0.3 11,-0.2 11,-0.2 -0.933 4.4 175.4-139.1 156.9 23.4 29.4 18.4 9 12 A F E -A 18 0A 31 9,-1.5 9,-1.8 -2,-0.3 2,-0.4 -0.967 13.3-160.9-160.6 151.3 24.4 27.6 15.3 10 13 A Y E -A 17 0A 36 -2,-0.3 2,-0.4 7,-0.2 7,-0.2 -0.994 16.5-156.0-136.8 132.1 27.2 25.3 14.1 11 14 A L E >> -A 16 0A 24 5,-3.2 4,-1.7 -2,-0.4 5,-1.3 -0.929 11.7-122.6-120.0 133.5 28.0 24.8 10.4 12 15 A N T 45S+ 0 0 54 -2,-0.4 -2, 0.0 2,-0.2 0, 0.0 -0.884 84.3 9.0-113.8 150.9 29.7 22.0 8.6 13 16 A P T 45S+ 0 0 114 0, 0.0 -1,-0.2 0, 0.0 -2, 0.0 -0.963 125.4 60.5 -86.5 8.5 32.0 21.6 6.8 14 17 A D T 45S- 0 0 66 2,-0.1 -2,-0.2 1,-0.1 3,-0.1 0.752 89.3-146.2 -64.6 -23.0 33.0 25.2 7.6
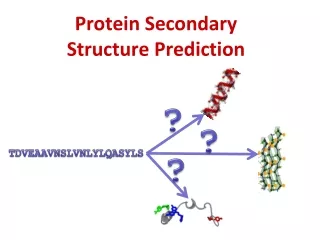
Q3 H E C Secondary Structure Prediction • What to predict? • All 8 types or pool types into groups DSSP * H = alpha helix * G = 310 -helix * I = 5 helix (pi helix) * E = extended strand * B = beta-bridge * T = hydrogen bonded turn * S = bend * C = coil
Q3 H E C Secondary Structure Prediction Straight HEC • What to predict? • All 8 types or pool types into groups * H = alpha helix * E = extended strand * T = hydrogen bonded turn * S = bend * C = coil * G = 310-helix * I = 5 helix (pi helix) * B = beta-bridge
Secondary Structure Prediction • Simple alignments • Align to a close homolog for which the structure has been experimentally solved. • Heuristic Methods (e.g., Chou-Fasman, 1974) • Apply scores for each amino acid an sum up over a window. • Neural Networks • Raw Sequence (late 80’s) • Blosum matrix (e.g., PhD, early 90’s) • Position specific alignment profiles (e.g., PsiPred, late 90’s) • Multiple networks balloting, probability conversion, output expansion (Petersen et al., 2000).
1974 Chou & Fasman ~50-53% 1978 Garnier 63% 1987 Zvelebil 66% 1988 Quian & Sejnowski 64.3% 1993 Rost & Sander 70.8-72.0% 1997 Frishman & Argos <75% 1999 Cuff & Barton 72.9% 1999 Jones 76.5% 2000 Petersen et al. 77.9% Improvement of accuracy
Simple Alignments • Solved structure of a homolog to query is needed • Homologous proteins have ~88% identical (3 state) secondary structure • If no close homologue can be identified alignments will give almost random results
Name P(a) P(b) P(turn) f(i) f(i+1) f(i+2) f(i+3) Ala 142 83 66 0.06 0.076 0.035 0.058 Arg 98 93 95 0.070 0.106 0.099 0.085 Asp 101 54 146 0.147 0.110 0.179 0.081 Asn 67 89 156 0.161 0.083 0.191 0.091 Cys 70 119 119 0.149 0.050 0.117 0.128 Glu 151 37 74 0.056 0.060 0.077 0.064 Gln 111 110 98 0.074 0.098 0.037 0.098 Gly 57 75 156 0.102 0.085 0.190 0.152 His 100 87 95 0.140 0.047 0.093 0.054 Ile 108 160 47 0.043 0.034 0.013 0.056 Leu 121 130 59 0.061 0.025 0.036 0.070 Lys 114 74 101 0.055 0.115 0.072 0.095 Met 145 105 60 0.068 0.082 0.014 0.055 Phe 113 138 60 0.059 0.041 0.065 0.065 Pro 57 55 152 0.102 0.301 0.034 0.068 Ser 77 75 143 0.120 0.139 0.125 0.106 Thr 83 119 96 0.086 0.108 0.065 0.079 Trp 108 137 96 0.077 0.013 0.064 0.167 Tyr 69 147 114 0.082 0.065 0.114 0.125 Val 106 170 50 0.062 0.048 0.028 0.053 Chou-Fasman propensities
Chou-Fasman • Generally applicable • Works for sequences with no solved homologs • But the accuracy is low! • The problem is that the method does not use enough information about the structural context of a residue
Neural Networks • Benefits • Generally applicable • Can capture higher order correlations • Inputs other than sequence information • Drawbacks • Needs a high amount of data (different solved structures). However, today nearly 7000 structures with low sequence identity/high resolution are solved • Complex method with several pitfalls
Weights Input Layer I K H E Output Layer E E C H V I I Q A E Hidden Layer Window IKEEHVIIQAEFYLNPDQSGEF….. Architecture
Sparse encoding Inp Neuron 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 AAcid A 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 R 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 N 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 D 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 C 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 Q 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 E 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 I 0 K 0 E 1 E 0 H 0 V 0 I 0 I 0 Q 0 A 0 E 0 0 0 0 0 0 Input Layer
A R N D C Q E G H I L K M F P S T W Y V B Z X * A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4 R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4 N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4 D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4 C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4 Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4 E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4 G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4 H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4 I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4 L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4 K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4 M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4 F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4 P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -2 -4 S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 -4 T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 0 -4 W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -2 -4 Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4 V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4 BLOSUM 62
-1 0 0 I 2 -4 K E 2 E 5 -2 H V 0 -3 I -3 I Q 1 -2 A -3 E -1 0 -1 -3 -2 -2 Input Layer
Weights Input Layer H E H C Output Layer E H C E C H E C Window Hidden Layer IKEEHVIIQAEFYLNPDQSGEF….. Secondary networks(Structure-to-Structure)
PHD method (Rost and Sander) • Combine neural networks with sequence profiles • 6-8 Percentage points increase in prediction accuracy over standard neural networks • Use second layer “Structure to structure” network to filter predictions • Jury of predictors • Set up as mail server
PSI-Pred (Jones) • Use alignments from iterative sequence searches (PSI-Blast) as input to a neural network • Better predictions due to better sequence profiles • Available as stand alone program and via the web
A R N D C Q E G H I L K M F P S T W Y V 1 I -2 -4 -5 -5 -2 -4 -4 -5 -5 6 0 -4 0 -2 -4 -4 -2 -4 -3 4 2 K -1 -1 -2 -2 -3 -1 3 -3 -2 -2 -3 4 -2 -4 -3 1 1 -4 -3 2 3 E 5 -3 -3 -3 -3 3 1 -2 -3 -3 -3 -2 -2 -4 -3 -1 -2 -4 -3 1 4 E -4 -3 2 5 -6 1 5 -4 -3 -6 -6 -2 -5 -6 -4 -2 -3 -6 -5 -5 5 H -4 2 1 1 -5 1 -2 -4 9 -5 -2 -3 -4 -4 -5 -3 -4 -5 1 -5 6 V -3 0 -4 -5 -4 -4 -2 -3 -5 1 -2 1 0 1 -4 -3 3 -5 -3 5 7 I 0 -2 -4 1 -4 -2 -4 -4 -5 1 0 -2 0 2 -5 1 -1 -5 -3 4 8 I -3 0 -5 -5 -4 -2 -5 -6 1 2 4 -4 -1 0 -5 -2 0 -3 5 -1 9 Q -2 -3 -2 -3 -5 4 -1 3 5 -5 -3 -3 -4 -2 -4 2 -1 -4 2 -2 10 A 2 -4 -4 -3 2 -3 -1 -4 -2 1 -1 -4 -3 -4 1 2 3 -5 -1 1 11 E -1 3 1 1 -1 0 1 -4 -3 -1 -3 0 3 -5 4 -1 -3 -6 -3 -1 12 F -3 -5 -5 -5 -4 -4 -4 -1 -1 1 1 -5 2 5 -1 -4 -4 -3 5 2 13 Y 3 -5 -5 -6 3 -4 -5 -2 -1 0 -4 -5 -3 3 -5 -2 -2 -2 7 1 14 L -1 -3 -4 -2 1 5 1 -1 -1 -1 1 -3 -3 1 -5 -1 -1 -2 3 -2 15 N -1 -4 4 1 5 -3 -4 2 -4 -4 -4 -3 -2 -4 -5 2 0 -5 0 0 16 P -2 4 -4 -4 -5 0 -3 3 2 -5 -4 0 -4 -3 0 1 -2 -1 5 -3 17 D -3 -2 1 5 -6 -2 2 2 -1 -2 -2 -3 -5 -4 -5 -1 2 -6 -3 -4 Position specific scoring matrices (PSI-BLAST profiles)
Sequence-to-structure • Window sizes 15,17,19 and 21 • Hidden units 50 and 75 • 10-fold cross validation => 80 predictions • Structure-to-structure • Window size 17 • Hidden units 40 • 10-fold cross validation => 800 predictions Several different architectures Output: C C H H C C C Output: C C C C C C C
Combining predictions from several networks improves the prediction • Combinations of 800 different networks were used in the method described by • Petersen TN et al. 2000, Prediction of protein secondary structure at 80 % accuracy. Proteins 41 17-20 The majority rules
Helix activities (output) Strand activities (output) Coil probabilities! (calculated) Coil conversion 0.05 0.1 0.15 … 1.0 0.05 0.99 0.10 0.15 0.9 0.83 0.75 . . . 1.0 Activities to probabilities
Benchmarking secondary structure predictions • EVA • Newly solved structures are send to prediction servers. • Every week http://cubic.bioc.columbia.edu/eva/sec/res_sec.html
EVA results (Rost et al., 2001) • PROFphd 77.0% • PSIPRED 76.8% • SAM-T99sec 76.1% • SSpro 76.0% • Jpred2 75.5% • PHD 71.7% • Cubic.columbia.edu/eva
Links to servers • Several links: http://cubic.bioc.columbia.edu/eva/doc/explain_methods.html#type_sec • ProfPHD http://www.predictprotein.org/ • PSIPRED http://bioinf.cs.ucl.ac.uk/psipred/ • JPred http://www.compbio.dundee.ac.uk/~www-jpred/ • SAM T02 http://www.cse.ucsc.edu/research/compbio/HMM-apps/T02-query.html
Practical Conclusions • If you need a secondary structure prediction use the newer methods based on advanced machine learning methods such as : • ProfPHD • PSIPRED • JPred • SAM T02 • And not one of the older ones such as : • Chou-Fasman • Garnier