Download

1 / 40
410 likes | 1.04k Views
Structure and stability of globular proteins. Aliphatic amino acids and Gly. Cyclic Imino Acid: Proline. Hydroxyl amino acids. Acidic amino acids. Amide amino acids. Basic amino acids. Histidine. Sulfur-containing amino acids. Aromatic amino acids.

E N D
Backbone conformation is described by φ and ψ angles. Picture from T. Przytycka, 2002
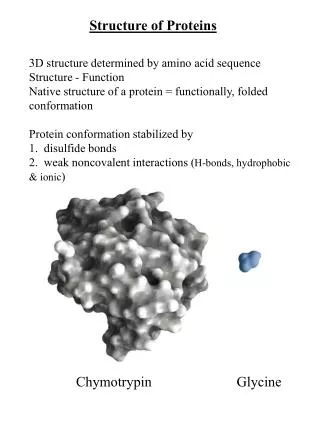
Protein folding.Anfinsen’s experiments. Assumption: amino acid sequence completely and uniquely determines the protein tertiary structure. Protein folding problem: find native conformation among the large number of alternative conformations. Ex: polypeptide chain of 100 residues can have ~ 9^100 different conformations.
Native proteins are marginally stable. Scale of interactions in proteins: - Interactions less than kT~0.6 kcal/mol are neglected. - ΔG ~ 5 - 20 kcal/mol Potential energy = Van der Waals + Electrostatic + … G U F ΔG Reaction coordinate
Electrostatic force. Coulomb’s law for two point charges in a vacuum: q – point charge, ε – dielectric constant ε = 2-3 inside the protein, ε = 80 in water Na+ Cl- d = 2.76 Å, E = 120 kcal/mol
Dipolar interactions. - 0.42 Dipole moment: O +0.42 C Interaction energy of two dipoles separated by the vector r: -0.20 N Peptide bond: μ = 3.5D, Water molecule: μ = 1.85D. +0.20 H
Van der Waals interactions. E (kcal/mol) Lennard-Jones potential: 0.2 repulsion 0 attraction - 0.2 2 4 6 8 10 12 Distance between centers of atoms
Van der Waals interactions. Three major types of interactions: • between two permanent dipoles • between a permanent and an induced dipole • between mutually induced dipoles
Hydrogen bonding patterns in globular proteins. 1. Most HB are local, close in sequence. 2. Most HB are between backbone atoms. 3. Most HB are within single elements of secondary structure. 4. Proteins are almost equally saturated by HB: 0.75 bond per amino acid.
Disulfide bonds. PROTEIN + GS-SG PROTEIN + GSHPROTEIN + 2GSH SH HS SH S-SG - Breakdown and formation of S-S bonds are catalyzed by disulfide isomerase. - In the cell S-S bonds are reversible, the energetic equilibrium is close to zero. - Secreted proteins have a lot of S-S bonds since outside the cell the equilibrium is shifted towards their formation.
Properties of water. O HB determines properties of water: - tetrahedral structure of ice, 4 HB neighbors, very open lattice. - liquid water forms HBonded “icebergs”. Unusual physical properties: - water expands when it freezes at 0 C. When ice is melted and then warmed, the liquid continues to contract upto 4C. - water has high melting and boiling points, large heat capacity. 0.957 Å 104.5° H H
Hydrophobic effect. H Hydrophobic interaction – tendency of nonpolar compounds to transfer from an aqueous solution to an organic phase. • The entropy of water molecules decreases when they make a contact with a nonpolar surface (TΔS = -9.6 kcal/mol for cyclohexane) . • The effect is entropic because the energy of HB is very high. • The hydrophobic effect is proportional to buried surface area, the energy is ~ 20-25 cal/mol/A^2 O H O H H
Protein folding: step by step. Disordered globule: hydrophobic – inside, hydrophilic - outside Extended chain Native highly ordered conformation
Hierarchy of protein structure. • Amino acid sequence • Secondary structure • Tertiary structure • Quaternary structure Picture from Branden & Tooze “Introduction to protein structure”
Right-handed alpha-helix. • Helix is stabilized by HB between backbone –NH and backbone carbonyl atom. • Geometrical characteristics: • 3.6 residues per turn • translation of 5.4 Å per turn • translation of 1.5 Å per residue
Loop regions are at the surface of protein molecules. Adjacent antiparallel β-strands are joined by hairpin loops. Loops are more flexible than helices and strands. Loops can carry binding and active sites, functionally important sites. Branden & Tooze “Introduction to protein structure”
Fibrous proteins. • Have extended structures, regular conformations, insoluble. • Basis for regularities in structure is in regularities in sequence. • Coiled coils (heptad repeats in sequence). • Collagen triple helix (every third residue is Gly).
Classwork I: CN3D viewer. • Go to http://ncbi.nlm.nih.gov • Select alpha-helical protein (hemoglobin) • Select beta-stranded protein (immunoglobulin) • Select multidomain protein 1I50, chain “A” • View them in CN3D
PDB databank. • Archive of protein crystal structures was established in 1971 with several structures. in 2002 – 17000 structures including NMR structures • Data processing: data deposition, annotation and validation • PDB code – nXYZ, n – integer, X, Y, Z -characters
Content of Data in the PDB. • Organism, species name • Full protein sequence • Chemical structure of cofactors and prosthetic groups • Names of all components of the structure • Qualitative description of the structural characteristics • Literature citations • Three-dimensional coordinates
Protein secondary structure prediction. Assumptions: • There should be a correlation between amino acid sequence and secondary structure. Short aa sequence is more likely to form one type of SS than another. • Local interactions determine SS. SS of a residues is determined by their neighbors (usually a sequence window of 13-17 residues is used). Exceptions: short identical amino acid sequences can sometimes be found in different SS. Accuracy: 65% - 75%, the highest accuracy – prediction of an α helix
Methods of SS prediction. • Chou-Fasman method • GOR (Garnier,Osguthorpe and Robson) • Neural network method
Chou-Fasman method. Amino acid itself determines the type of secondary structure, which it is likely to adopt. Analysis of frequences for all amino acids to be in different types of SS. Ala, Glu, Leu and Met – strong predictors of alpha-helices, Pro and Gly predict to break the helix. Prediction of an alpha-helix - if four of six adjacent amino acids have high probability to be a helix. Prediction of a beta-strand – if three of five adjacent amino acids have high probability to be a strand.
GOR method. Assumption:formation of SS of an amino acid is determined by the neighboring residues (usually a window of 17 residues is used). GOR uses principles of information theory for predictions. Method maximizes the information difference between two competing hypotheses: that residue “a” is in structure “S”, and that “a” is not in conformation “S”.
Neural network method. Input layer Input sequence window Output layer Predicted SS Hidden layer L A W P G E V G A S T Y P α Si Hj Oi 1 β 0 coil 0 WijSj HjOi
PHD – neural network program with multiple sequence alignments. • Blast search of the input sequence is performed, similar sequences are collected. • Multiple alignment of similar sequences is used as an input to a neural network. • Sequence pattern in multiple alignment is enhanced compared to if one sequence used as an input.
Classwork • Go to http://ncbi.nlm.nih.gov, search for protein “flavodoxin” in Entrez, retrieve its amino acid sequence. • Go to http://cubic.bioc.columbia.edu/predictprotein and run PHD on the sequence.