Download
1 / 1
20 likes | 132 Views
A CONVENIENT FORMATION OF HIGHLY FUNCTIONALIZED 5-BROMO-1,3-THIAZOLES Eduard Doluši ć , Sara Modaffari, Johan Wouters, Bernard Masereel and Raphaël Frédérick NAmur MEDIcine Center (NAMEDIC), NARILIS, 61 rue de Bruxelles, B-5000 Namur, Belgium. NAMEDIC. 1. Introduction.

E N D
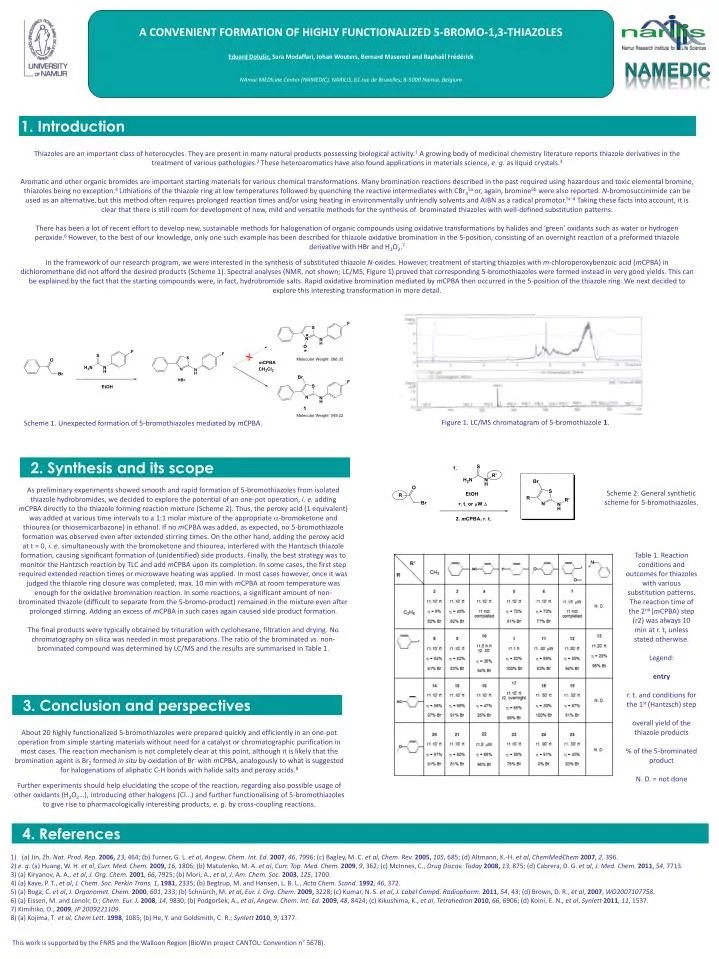
A CONVENIENT FORMATION OF HIGHLY FUNCTIONALIZED 5-BROMO-1,3-THIAZOLES Eduard Dolušić, Sara Modaffari, Johan Wouters, Bernard Masereel and Raphaël Frédérick NAmur MEDIcine Center (NAMEDIC), NARILIS, 61 rue de Bruxelles, B-5000 Namur, Belgium NAMEDIC 1. Introduction Thiazoles are an important class of heterocycles. They are present in many natural products possessing biological activity.1 A growing body of medicinal chemistry literature reports thiazole derivatives in the treatment of various pathologies.2 These heteroaromatics have also found applications in materials science, e. g. as liquid crystals.3 Aromatic and other organic bromides are important starting materials for various chemical transformations. Many bromination reactions described in the past required using hazardous and toxic elemental bromine, thiazoles being no exception.4 Lithiations of the thiazole ring at low temperatures followed by quenching the reactive intermediates with CBr45a or, again, bromine5b were also reported. N-bromosuccinimide can be used as an alternative,but this method often requires prolonged reaction times and/or using heating in environmentally unfriendly solvents and AIBN as a radical promotor.5c-d Taking these facts into account, it is clear that there is still room for development of new, mild and versatile methods for the synthesis of brominated thiazoles with well-defined substitution patterns. There has been a lot of recent effort to develop new, sustainable methods for halogenation of organic compounds using oxidative transformations by halides and ‘green’ oxidants such as water or hydrogen peroxide.6 However, to the best of our knowledge, only one such example has been described for thiazole oxidative bromination in the 5-position, consisting of an overnight reaction of a preformed thiazole derivative with HBr and H2O2.7 In the framework of our research program, we were interested in the synthesis of substituted thiazole N-oxides. However, treatment of starting thiazoles with m-chloroperoxybenzoic acid (mCPBA) in dichloromethane did not afford the desired products (Scheme 1). Spectral analyses (NMR, not shown; LC/MS, Figure 1) proved that corresponding 5-bromothiazoles were formed instead in very good yields. This can be explained by the fact that the starting compounds were, in fact, hydrobromide salts. Rapid oxidative bromination mediated by mCPBA then occurred in the 5-position of the thiazole ring. We next decided to explore this interesting transformation in more detail. Figure 1. LC/MS chromatogram of 5-bromothiazole 1. Scheme 1. Unexpected formation of 5-bromothiazoles mediated by mCPBA. 2. Synthesis and its scope As preliminary experiments showed smooth and rapid formation of 5-bromothiazoles from isolated thiazole hydrobromides, we decided to explore the potential of an one-pot operation, i. e. adding mCPBA directly to the thiazole forming reaction mixture (Scheme 2). Thus, the peroxy acid (1 equivalent) was added at various time intervals to a 1:1 molar mixture of the appropriate a-bromoketone and thiourea (or thiosemicarbazone) in ethanol. If no mCPBA was added, as expected, no 5-bromothiazole formation was observed even after extended stirring times. On the other hand, adding the peroxy acid at t = 0, i. e. simultaneously with the bromoketone and thiourea, interfered with the Hantzsch thiazole formation, causing significant formation of (unidentified) side products. Finally, the best strategy was to monitor the Hantzsch reaction by TLC and add mCPBA upon its completion. In some cases, the first step required extended reaction times or microwave heating was applied. In most cases however, once it was judged the thiazole ring closure was completed, max. 10 min with mCPBA at room temperature was enough for the oxidative bromination reaction. In some reactions, a significant amount of non-brominated thiazole (difficult to separate from the 5-bromo-product) remained in the mixture even after prolonged stirring. Adding an excess of mCPBA in such cases again caused side product formation. The final products were typically obtained by trituration with cyclohexane, filtration and drying. No chromatography on silica was needed in most preparations. The ratio of the brominated vs. non-brominated compound was determined by LC/MS and the results are summarised in Table 1. Scheme 2. General synthetic scheme for 5-bromothiazoles. Table 1. Reaction conditions and outcomes for thiazoles with various substitution patterns. The reaction time of the 2nd (mCPBA)step(r2)was always 10 min at r. t, unless stated otherwise. Legend: entry r. t. and conditions for the 1st (Hantzsch) step overall yield of the thiazole products % of the 5-brominated product N. D. = not done 3. Conclusion and perspectives About 20 highly functionalized 5-bromothiazoles were prepared quickly and efficiently in an one-pot operation from simple starting materials without need for a catalyst or chromatographic purification in most cases. The reaction mechanism is not completely clear at this point, although it is likely that the bromination agent is Br2 formed in situ by oxidation of Br- with mCPBA, analogously to what is suggested for halogenations of aliphatic C-H bonds with halide salts and peroxy acids.8 Further experiments should help elucidating the scope of the reaction, regarding also possible usage of other oxidants (H2O2…), introducing other halogens (Cl…) and further functionalising of 5-bromothiazoles to give rise to pharmacologically interesting products, e. g. by cross-coupling reactions. 4. References • (a) Jin, Zh. Nat. Prod. Rep. 2006,23, 464; (b) Turner, G. L. et al, Angew. Chem. Int. Ed. 2007, 46, 7996; (c) Bagley, M. C. et al, Chem. Rev. 2005,105, 685; (d) Altmann, K.-H. et al, ChemMedChem 2007, 2, 396. • 2) e. g.(a) Huang, W. H. et al, Curr. Med. Chem. 2009,16, 1806; (b) Matulenko, M. A. et al, Curr. Top. Med. Chem. 2009, 9, 362; (c) McInnes, C., Drug Discov. Today 2008,13, 875; (d) Cabrera, D. G. et al, J. Med. Chem. 2011, 54, 7713. • 3) (a) Kiryanov, A. A., et al, J. Org. Chem.2001, 66, 7925; (b) Mori, A., et al, J. Am. Chem. Soc. 2003, 125, 1700. • 4) (a) Kaye, P. T., et al, J. Chem. Soc. Perkin Trans. 1, 1981, 2335; (b) Begtrup, M. and Hansen, L. B. L., Acta Chem. Scand. 1992, 46, 372. • 5) (a) Boga, C. et al, J. Organomet. Chem. 2000, 601, 233; (b) Schnürch, M. et al, Eur. J. Org. Chem. 2009, 3228; (c) Kumar, N. S. et al, J. Label Compd. Radiopharm.2011, 54, 43; (d) Brown, D. R., et al, 2007, WO2007107758. • 6) (a) Eissen, M. and Lenoir, D.; Chem. Eur. J.2008, 14, 9830; (b) Podgoršek, A., et al, Angew. Chem. Int. Ed. 2009, 48, 8424; (c) Kikushima, K., et al, Tetrahedron 2010, 66, 6906; (d) Koini, E. N., et al, Synlett 2011, 11, 1537. • 7) Kimihiko, O., 2009, JP 2009221109. • 8) (a) Kojima, T. et al, Chem Lett. 1998, 1085; (b) He, Y. and Goldsmith, C. R.; Synlett2010, 9, 1377. This work is supported by the FNRS and the Walloon Region (BioWin project CANTOL: Convention n° 5678).