Download

1 / 1
10 likes | 173 Views
A MULTIBODY ATOMIC STATISTICAL POTENTIAL FOR PREDICTING ENZYME-INHIBITOR BINDING ENERGY Majid Masso (mmasso@gmu.edu) Laboratory for Structural Bioinformatics, School of Systems Biology, George Mason University, 10900 University Blvd. MS 5B3, Manassas, Virginia 20110, USA.

E N D
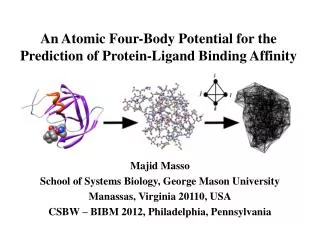
A MULTIBODY ATOMIC STATISTICAL POTENTIAL FOR PREDICTING ENZYME-INHIBITOR BINDING ENERGY Majid Masso (mmasso@gmu.edu) Laboratory for Structural Bioinformatics, School of Systems Biology, George Mason University, 10900 University Blvd. MS 5B3, Manassas, Virginia 20110, USA II. Protein Data Bank (http://www.rcsb.org/pdb) III. Macromolecular Modeling I. Abstract Accurate prediction of enzyme-inhibitor binding energy has the capacity to speed drug design and chemical genomics efforts by helping to narrow the focus of experiments. Here a non-redundant set of three hundred high-resolution crystallographic enzyme-inhibitor structures was compiled for analysis, complexes with known binding energies (ΔG) based on the availability of experimentally determined inhibition constants (ki). Additionally, a separate set of over 1400 diverse high-resolution macromolecular crystal structures was collected for the purpose of creating an all-atom knowledge-based statistical potential, via application of the Delaunay tessellation computational geometry technique. Next, two hundred of the enzyme-inhibitor complexes were randomly selected to develop a model for predicting binding energy, first by tessellating structures of the complexes as well as the enzymes without their bound inhibitors, then by using the statistical potential to calculate a topological score for each structure tessellation. We derived as a predictor of binding energy an empirical linear function of the difference between topological scores for a complex and its isolated enzyme. A correlation coefficient (r) of 0.79 was obtained for the experimental and calculated ΔG values, with a standard error of 2.34 kcal/mol. Lastly, the model was evaluated with the held-out set of one hundred complexes, for which structure tessellations were performed in order to calculate topological score differences, and binding energy predictions were generated from the derived linear function. Calculated binding energies for the test data also compared well with their experimental counterparts, displaying a correlation coefficient of r = 0.77 with a standard error of 2.50 kcal/mol. • PDB – repository of solved (x-ray, nmr, ...) structures • Each structure file contains atomic 3D coordinate data • Native structure is conformation having lowest energy • Physics-based energy calculations using quantum mechanics are computationally impractical • Same for molecular mechanics-based potential energy functions (i.e., force fields): E(total) = E(bond) + E(angle) + E(dihedral) + E(electrostatic) + E(van der Waals) • Alternative (our approach): knowledge-based potentials of mean force (i.e., generated from known protein structures) Atom X Y Z : : : : IV. Knowledge-Based Potentials of Mean Force V. Motivational Example:Pairwise Amino Acid Potential VI. All-Atom Four-Body Statistical Potential • Obtain diverse PDB dataset of 1417 single chain and multimeric proteins, many complexed to ligands (see XV. References) • Six-letter atomic alphabet: C, N, O, S, M (metals), X (other) • Apply Delaunay tessellation to the atomic point coordinates of each PDB file – objectively identifies all nearest-neighbor quadruplets of atoms in the structure (8 angstrom cutoff) • Assumptions: • At equilibrium, native state has global free energy min • Microscopic states (i.e., features) follow Boltzmann dist • Examples: • Well-documented in the literature: distance-dependent pairwise interactions at the atomic or amino acid level • This study: inclusion of higher-order contributions by developing all-atom four-body statistical potentials • Motivation (our prior work): • Four-body protein potential at the amino acid level • A 20-letter protein alphabet yields 210 residue pairs • Obtain large, diverse PDB dataset of single protein chains • For each residue pair (i, j), calculate the relative frequency fij with which they appear within a given distance (e.g., 12 angstroms) of each other in all the protein structures • Calculate a rate pij expected by chance alone from a background or reference distribution (more later…) • Apply inverted Bolzmann principle: sij = log(fij / pij) quantifies interaction propensity and is proportional to the energy of interaction (by a factor of ‘–RT’) VII. All-Atom Four-Body Statistical Potential VIII. Summary Data for the 1417 Structure Files and their Delaunay Tessellations IX. All-Atom Four-Body Statistical Potential • A six-letter atomic alphabet yields 126 distinct quadruplets • For each quad (i, j, k, l), calculate observed rate of occurrence fijklamong all tetrahedra from the 1417 structure tessellations • Compute rate pijkl expected by chance from a multinomial reference distribution: • an = proportion of atoms from all structures that are of type n • tn = number of occurrences of atom type n in the quad • Apply inverted Bolzmann principle: sijkl = log(fijkl / pijkl) quantifies the interaction propensity and is proportional to the energy of atomic quadruplet interaction X. Topological Score (TS) XII. Application of ΔTS: Predicting Enzyme–Inhibitor Binding Energy XI. Topological Score Difference (ΔTS) • Delaunay tessellation of any macromolecular structure yields an aggregate of tetrahedral simplices • Each simplex can be scored using the all-atom four-body potential based on the quad present at the four vertices • Topological score (or ‘total potential’) of the structure: the sum of all constituent simplices in the tessellation • MOAD – repository of exp. inhibition constants (ki) for protein–ligand complexes whose structures are in PDB • Collected ki values for 300 complexes reflecting diverse protein structures • Obtained exp. binding energy from ki via ΔGexp= –RTln(ki) • Calculated ΔTS for complexes TS = Σsijkl sijkl XIII. Predicting Enzyme–Inhibitor Binding Energy XIV. Predicting Enzyme–Inhibitor Binding Energy XV. References and Acknowledgments • PDB dataset: http://proteins.gmu.edu/automute/tessellatable1417.txt • Train/test dataset: http://proteins.gmu.edu/automute/MOAD300ki.txt • PDB (structure DB): http://www.rcsb.org/pdb • MOAD (ligand binding DB): http://bindingmoad.org/ • Qhull (Delaunay tessellation): http://www.qhull.org/ • UCSF Chimera (ribbon/ball-stick structure visualization): http://www.cgl.ucsf.edu/chimera/ • Matlab (tessellation visualization): http://www.mathworks.com/products/matlab/ • For the test set of 100 remaining complexes: • r = 0.77 between ΔGcalc and ΔGexp • SE = 2.50 kcal/mol • Fitted regression line is y = 1.07x + 0.46 • All training/test data available online as a text file (see XV. References) • Randomly selected 200 complexes to train a model • Correlation coefficient r = 0.79 between ΔTS and ΔGexp • Empirical linear transform of ΔTS to reflect energy values: • ΔGcalc = (1 / 0.0003) × ΔTS – 6.24 • Linear => same r = 0.79value between ΔGcalc and ΔGexp • Also, standard error of SE = 2.34 kcal/mol and fitted regression line of y = 0.98x – 0.41 (y = ΔGcalc and x = ΔGexp)