Download

1 / 24
450 likes | 1.45k Views
Hydrogenation. Textbook H: Chapter 15.1 – 15.6 Textbook A: Chapter 14.1 – 14.2. Hydrogenation. All mechanisms involve metal hydrides. Oxidative addition Sigma-bond metathesis Heterolytic activation. Wilkinson’s catalyst: OA of H 2 (hydride mechanism).

E N D
Hydrogenation Textbook H: Chapter 15.1 – 15.6 Textbook A: Chapter 14.1 – 14.2
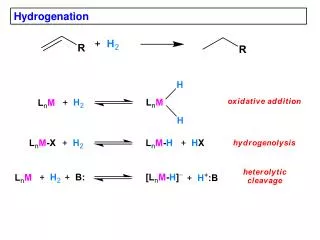
Hydrogenation • All mechanisms involve metal hydrides. • Oxidative addition • Sigma-bond metathesis • Heterolytic activation
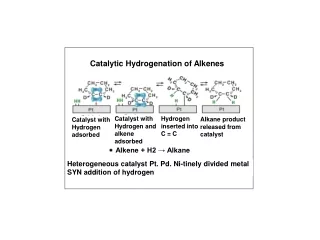
Wilkinson’s catalyst: OA of H2 (hydride mechanism) H2 adds to the catalyst before the olefin. The last step of the catalytic cycle is irreversible. This is very useful because a kinetic product ratio can be obtained. Simplistically, the relative rates suggest that the rate-determining step is OA of H2.
Cationic catalysts: OA of H2 (olefin mechanism) Cationic Rh complexes: Schrock and Osborn Cationic Ir complexes: Crabtree With cationic catalysts, because the metal center is a strong Lewis acid, the olefin coordinates first and then the oxidative addition of H2 occurs.
Sigma-bond metathesis It is found for early TM, lanthanides, and actinides (d0fn configuration).
Heterolytic H2 activation • The protons of a dihydrogen ligand are more acidic than those in free H2: many H2 complexes can be deprotonated with NEt3. • Once H2 activation takes place, the rest of the catalytic cycle is similar to that for Wilkinson’s catalyst. • RuCl2(PPh3)2 hydrogenates selectively terminal double bonds over internal double bonds:
H-transfer reactions Transfer hydrogenation avoids the use of high pressures of H2. The ketone substrate does not coordinate to the metal, but is oriented in the second-coordination sphere by forming hydrogen bonds. Reference: Morris, R. H. et al. Coord. Chem. Rev. 2004, 248, 2201
Homolytic H2 activation A very early example of a homogeneous hydrogenation catalyst (Iguchi, 1942): The resulting organic radical needs to be moderately stable: only “activated” alkenes will be hydrogenated (formation of a conjugated radical).
Asymmetric hydrogenation The intermediate alkene adduct observed in the NMR spectra is not the one leading to the major product. Enantioselectivity is determined in the first irreversible step after the enantiocenter is formed (not always the rate-determining step).
Alkene isomerization: hydride mechanism It requires an M-H bond and a vacant site. It is often a side reaction. • All steps are reversible: TD ratio is formed if catalyst is active long enough. • Insertion to give the 1° alkyl is favored for many catalysts: nonproductive cycling. • The initial cis/trans ratio depends on the catalyst; the cis isomer is often favored. In the final • product mixture, the trans isomer is predominant.
Alkene isomerization: allyl mechanism It is found for metal fragments that have 2 x 2e vacant sites but no hydrides: Fe(CO)3. Crossover experiment to distinguish between allyl and hydride isomerization mechanisms:
Olefin additions Textbook H: Chapter 16.1 – 16.5 Textbook A: Chapter 14.3 – 14.6
Alkene additions With transition metal catalysts the control of selectivity is not simply explained by the stability of the carbocation intermediate: anti-Markovnikov products are possible. Reference: Beller, M. et al.Angew. Chem. Int. Ed. 2004, 43, 3368
Hydrocyanation of ethylene The reductive elimination (rate-determining step) is promoted by the addition of electron-withdrawing phosphite ligands. If styrene is used instead of ethylene, a benzyl intermediate forms.
Hydrocyanation of butadiene Initial addition of HCN leads to 1:2 in a 1:2 ratio, but they equilibrate to 1:9 via the retro-reaction (involves C-C breaking, but the allyl cyanide species is stable). Although 3 is more stable than 4, formation of 4 is kinetically controlled. Lewis acids are added as cocatalysts. They accelerate the reductive elimination (formation of 5), probably by complexation to CN, thus decreasing the electron density at the metal. Adiponitrile (5) is a precursor to 1,6-hexanediamine, one of the components of 6,6-nylon and polyurethanes.
Hydroboration The alkene could insert either in the Rh-H or the Rh-B bond (both are cis to the coordinated olefin). Data supporting both pathways have been reported. Reference: Crudden, C. M.; Edwards, D.Eur. J. Org. Chem. 2003, 24, 4695
Hydrosilylation Hydrosilylation can be applied to alkenes, alkynes, and carbonyl compounds. There are only few examples of selective dehydrogenative silylations. Platinum catalysts for hydrosilylation (used in industry): Although molecular catalysts are proposed for Rh, Ru, Ir, Pd, large clusters or small colloids are active for Pt. Reference: Marciniec, B.Silicon Chemistry 2002, 1, 155
Chalk-Harrod mechanism for hydrosilylation The two side products, vinylsilane and alkane are always formed in a 1:1 ratio.
Asymmetric palladium catalysts Catalytic asymmetric hydrosilylation of alkenes is a useful tool for the synthesis of optically active alcohols. Unlike all other metal catalysts, Pd catalysts give products with the SiR3 group at the secondary carbon. This selectivity is determined by the steric constraints of the ligand. The X (OMe) group has little influence on the course of the reaction and it is not coordinated during the catalytic cycle.
Iron catalysts for alkene hydrosilylation A M Tondreau, P. J. Chirik et al. Science 2012, 335, 567-570 Published by AAAS
Early transition metals and lanthanides Hydroamination was first developed with Ln catalysts. Ln complexes work best in intramolecular versions. Alkynes are more reactive than alkenes.
Late TMs: oxidative amination a The overall mechanism for the Pd-catalyzed oxidation of olefins. b C–heteroatom bond-forming by nucleophilic attack on a coordinated olefin c Migratory insertion into an M–heteroatom bond John F. Hartwig - Nature 455, 314-322(18 September 2008)
Relative rates of olefin insertion into alkyl, amide and alkoxo complexes Rationalization for why the rates of olefin insertion into late TM alkoxides and amides are faster than into late TM alkyls, based on the destabilization of the alkoxo and amide reactants and the stabilization of the products of insertion into the alkoxo and amido complexes by an M–Y dative bond John F. Hartwig - Nature 455, 314-322(18 September 2008)
Recently discovered reactions of transition-M–heteroatom bonds a RE to form C–N, C–O and C–S bonds in amines, ethers, and thioethers b OA of amine N–H bonds c Migratory insertions of olefins into metal amides and metal alkoxides d [2 + 2] Cycloadditions between olefins and M–imido or M–oxo complexes. These reactions are analogues of classic reactions occurring at M–C bonds and have only recently been discovered. John F. Hartwig - Nature 455, 314-322(18 September 2008)