Download

1 / 57
570 likes | 721 Views
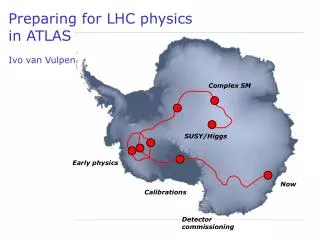
Now-16.12.04. Genome based views of organisms. Genomics: READING genome sequences ASSEMBLY of the sequence ANNOTATION of the sequence. carry out dideoxy sequencing ( Sanger ). connect seqs. to make whole chromosomes. find the genes!. Reading:. DNA target sample. SHEAR. Reads.

E N D
Genome based views of organisms Genomics: READING genome sequences ASSEMBLY of the sequence ANNOTATION of the sequence carry out dideoxy sequencing (Sanger) connect seqs. to make whole chromosomes find the genes!
Reading: DNA target sample SHEAR Reads LIGATE & CLONE Primer SEQUENCE Vector Shotgun DNA Sequencing of whole genome (WGS)
Reading: DNA samples in bacteria are picked, processed & sequenced robotically, -many thousands per day in large sequencing centers http://itgmv1.fzk.de/www/itg/uetz/robot_movie.html
We depend on Sanger dideoxy – sequencing Reading: Movie MCB0701_loop
Reading: Dideoxy sequencing done robotically…Hi-throughput
Genomics: READING genome sequences ASSEMBLY of the sequence ANNOTATION of the sequence Robotically do dideoxy-dye data collection connect seqs. to make whole chromosomes
Assembly: End Reads (Mates) Central steps of the assembly Primer ......................................ctgccaatgggc 12 |||||||||||| 401 caatcggccatggtggcatcgatcgcggcggcgttggactgccaatgggc 450 . . . . . 13 gctggcacgatgcccggcggtgtgggcggtggaccggggggcgtggccgg 62 |||||||||||||||||||||||||||||||||||||||||||||||||| 451 gctggcacgatgcccggcggtgtgggcggtggaccggggggcgtggccgg 500 . . . . . 63 tggaggaccccaggtgggagtcggtccacccgggagcggtaacggtggca 112 |||||||||||||||||||||||||||||||||||||||||||||||||| 501 tggaggaccccaggtgggagtcggtccacccgggagcggtaacggtggca 550 Smith-Waterman type alignment, >96%
Assembly: Human repeats make assembly difficult 50% of human genome is (dead)transposons/mobile elements!! 50% of human genome is (dead) transposons/mobile elements!!
Assembly: DNA target sample SHEAR SIZE SELECT End Reads (Mates) LIGATE & CLONE Primer SEQUENCE Vector HUMAN Shotgun DNA Sequencing pair-mates 2,000 Base pair library 10,000 Base pair library 150,000 Base pair library
Assembly: Each nucleotide sequenced many times Assembly Progression(Macro View)
Assembly: Labour intensive “Genome closure”
Assembly: The challenge of eukaryotic genomes E. coli Genome 4 million bp The Human Genome 3 billion bp 50% of genome is repeat sequences!
Assembly:There is a second strategy for genome sequencing All new projects now do WGS (whole genome shotgun) approach But…many genomes were donein the past by ordered clone based sequencing As a user of genome databases, you’ll find many sequenced organisms whose data reflect the ordered clone approach
Assembly:Strategy II: Ordered clone based sequencing ~300,000 bp
Assembly: The two alternative genome sequencing strategies 2. Whole Genome Shotgun approach • Ordered clone • approach
Annotation: Genomics: READING genome sequences ASSEMBLY of the sequence ANNOTATION of the sequence Robotically do dideoxy-dye data collection Whole genome shotgun OR Ordered clones find the genes !
Annotation: Genomics: READING genome sequences ASSEMBLY of the sequence ANNOTATION of the sequence find the genes ! • ab initio • by evidence
Annotation: For Bacterial genomes, ab initio is adequate ab initio: “from the beginning” יש מאין from first principles… ORFs are MOST of prokaryotic genome
Annotation: ab initio – finding ORFs • -85-88% of the nucleotides are associated with coding sequence • in the bacterial genomes that have been completely sequenced. • example: in Escherichia coli there are 4288 genes that • have an average of 950 bp of coding sequence • and are separated by an average of just 118 bp. So first, to find genes in prokaryotic DNA, search for ORFs!!
Annotation: ab initio – finding ORFs Scanning Prokaryotic DNA for ORF = stop = start (methionine)
Annotation: ab initio – finding ORFs Human and Yeast codon usage
Annotation: ab initio – evaluating ORFs Scanning Prokaryotic Codons for Usage Profile (Same gene [LacZ] as seen in previous slide)
Annotation: ab initio – beyond ORFs beyond ORFs: • -Prokaryotes have short, simple promoters that are • easy to recognize • -Transcriptional terminators often consist of short inverted • repeats followed by a run of Ts. • -Therefore, programs that find prokaryotic genes search for: • ORFs 60 or more codons long –and codon usage • promoters at the 5' end • Terminators at the 3' end • Homology to known genes from other prokaryotes • Shine-Dalgarno sequences • `
Annotation: ab initio – automated Prokaryotic gene finder examples Glimmer- Interpolated Markov Model method GrailII- Neural Network method (See BioInfo text – Fig 8.8)
Annotation: results
Annotation: Multicellular eukaryotes
Annotation: Multicellular eukaryotes
Annotation: Multicellular eukaryotes
Annotation: 2 ways to annotate eukaryotic genomes: -ab initio gene finders: Work on basic biological principles: Open reading frames Codon usage Consensus splice sites Met start codons ….. -Genes based on previous knowledge….EVIDENCE -cDNA sequence of the gene’s message -cDNA of a closely related gene’ message sequence -Protein sequence of the known gene Same gene’s Same gene’s from another species Related gene’s protein……. -ab initio gene finders: Work on basic biological principles: Open reading frames Codon usage Consensus splice sites Met start codons ….. Genes based on previous knowledge-EVIDENCE -cDNA sequence of the gene’s message -cDNA of a related gene’s message seq. -Protein sequence of the known gene Same gene’s Same gene’s from another species Related gene’s protein…….
[t]BLAST[x/n/p] t : Translate a DNA database in all 6 reading frames for comparison with a Protein query. x : Translate a nucleotide query in all 6 reading frames for comparison with a Protein database. p : Comparison is against a Protein database. n : Comparison is against a Nucleotide database. BLAST Versions of the program
[t]BLAST[x/n/p] t : Translate a DNA database in all 6 reading frames for comparison with a Protein query. x : Translate a nucleotide query in all 6 reading frames for comparison with a Protein database. p : Comparison is against a Protein database. n : Comparison is against a Nucleotide database. BLAST Versions of the program tBLASTn BLASTn nucleotide vs. nucleotide
Annotation: Evidence based A Single Gene of Interest Annotation Hundreds of Gene Families Thousands of Genes Millions of Blast Hits Tens of Scaffolds Thousands of Contigs Millions of Reads
Annotation: Evidence based Human Genome: Y2K-2000 “What was annotated (initial pass)?” PrimarySecondary (Evidence) (from same species) Repetitive DNAs Matches: tRNAs ESTs CpG islands Full length cDNAs Transcripts (including alternatives) Protein Similarity Transcription factor binding sites BLASTn BLASTn BLASTn BLASTn tBLASTn tBLASTX
Annotation: Evidence based From human pipeline to autoannotation ASSEMBLED SEQUENCE Precomputes Repeat Masker BlastX BlastN tRNAScan CpG Gene finders nraa CHGI Genscan CRGI fgenesH dBEST (subset) Grail-exp Various mRNAs dBSTS mouse fragments AUTOANNOTATION Feature Clustering and Resolution
Annotation: Annotation: Evidence based Transcription Unit
start and stop site predictions Unique identifiers Splice site predictions Homology based exon predictions computational exon predictions Tracking information Consensus gene structure (both strands)