Download

1 / 99
1k likes | 1.2k Views
Identification of regulatory elements using high-throughput binding evidence. Inference of population structure on large genetic data sets. Stoyan Georgiev advisors: Uwe Ohler and Sayan Mukherjee Computational Biology and Bioinformatics, Duke University February 2011. Outline.

E N D
Identification of regulatory elements using high-throughput binding evidence. Inference of population structure on large genetic data sets. StoyanGeorgiev advisors: Uwe Ohler and Sayan Mukherjee Computational Biology and Bioinformatics, Duke University February 2011
Outline • Motif analysis • Transcriptional regulation • genome-wide DNA binding data (Georgiev et al. 2010) • Post-transcriptional regulation • transcriptome-wide RNA binding data (Mukherjee et al., under review; Corcoran* and Georgiev* et al., submitted) • Inference of population structure • randomized algorithm
Outline • Introduction • Transcriptional regulation • Problem statement • Genomic assays • Statistical framework • Results • Post-transcriptional regulation
Gene regulation DNA motifs miRBP RBP RNA-binding Proteins Transcription Splicing, Capping, Poly-adenylation Nucleus Export Cytoplasm Stability Translation miR-RBP complexes RNA motifs
Gene regulatory code • Transcriptional regulation: short patterns in DNA (motifs) control the initiation of production of gene transcripts • mechanism: sequence-specific DNA binding proteins (TFs) Motif Discovery Tool: cERMIT (Georgiev et al. 2010) • Post-transcriptional regulation: short patterns in RNA control the utilization of gene transcripts • mechanism: sequence-specific RNA binding proteins (RBPs), or microRNA mediated Motif Analysis Tool (Corcoran* and Georgiev* et al.; Mukherjee et al.)
Transcriptional regulation • Chromatin arrangement • Activity of transcription factors • intra-cellular environment • cis-regulatory code • DNA methylation • Copy Number Variation
location Simplified abstraction
cERMIT • Computational tool for de-novo motif discovery • Predict binding motif and functional targets of a specific transcription factor of interest (e.g. TF) using genome-wide measurements of binding (e.g. ChIP-seq, ChIP-chip) (Georgiev et al. 2010) • Input: set of sequence regions with assigned binding evidence • Output: ranked list of predicted binding motifs and corresponding target locations
Brief introduction to cERMIT • Binding site representation: consensus sequence • Search for the "best" binding site that explains the genome-wide binding evidence. • "best“: occurs in regions that tend to have high evidence of being bound (this is formalized as a normalized average score) • can evaluate all possible binding sites up to some reasonable length...in theory • in practice, we try to cover as many as possible • start with all possible 5-mers (AAAAA, AAAAG, AAAAC,...,TTTTT) • for each, evaluate its "neighbours“ and replace it with the "best" one • repeat until no neighbour scores better than the current motif
sequence regions sequence regions sequence regions high evidence AAAAA AAAAG AAAAC AAAAT AAAGA . . . . . TTTCT TTTTA TTTTG TTTTC TTTTT RTGASTCA TGACTCA RTGASTCAK GAWTCAYY TGACTCA TGAWTCAK . . . . . ES = 15.0 ES = 1.5 low evidence evolved motifs 512 seed motifs Algorithmic view ES = normalized average binding evidence
ChIP-seq motif discovery input output
ChIP-chip validation • conservation filter improves prediction accuracy (Georgiev et al. 2010)
Example yeast ChIP-chip output GCN4 SKO1
CTCF STAT1 SRF Human ChIP-seq prediction literature Barski et al. 2007 Robertson et al. 2007 Valouev et al. 2008
Gene regulatory code • Post-transcriptional regulation: short patterns in RNA control the utilization of gene transcripts • mechanism: sequence-specific RNA binding proteins (RBPs), or microRNA mediated to control translation Motif Analysis Tool (Corcoran*, Georgiev* et al.; Mukherjee et al.)
PAR-CLIP • CLIP: Cross linking and immunoprecipitation • a method of transcriptome-wide identification of RNA-protein interaction sites – problem, quite noisy • PAR-CLIP = CLIP + photoactivatable nucleotides • more efficient cross linking • directly observable evidence of Protein-RNA cross linking: upon reverse transcription T->C conversion near or at the interaction site
PAR-CLIP 1. culture with 4-SU 2. cross-link 3. Immunoprecipitate & size-select 4. convert into a cDNA library & sequence [Hafner et al. 2010]
RBP motif analysis pipeline RBP cERMIT Motif predictions Motif seeds
predicted motif Pumilio • 2 million mapped reads • # clusters with site / total # clusters = 1,162 / 8,483 (Hafner et al. 2010)
Summary • cERMIT: motif discovery using genome-wide binding data • identify motifs that are highly enriched in targets with high binding evidence. • applicable to RNA and DNA binding data • adjust for sequence biases and other potential confounders using linear regression framework • In progress… • Bayesian formulation • improve stability of predictions • more comprehensive search
Inference of population structure and generalized eigendecomposition
Outline • Motivation • Current approaches • Extensions • large data sets • supervised dimension reduction • Empirical results • Wishart simulation • WCCC Crohn’s disease data set
Motivation A classic problem in biology and genetics is to study population structure (Cavalli-Sforza 1978, 2003) Genotype data on millions of loci and thousands of individuals Can we detect structure based on the genetic data? infer population demographic histories correct for population structure in disease association studies correspondence to geography
Current approaches Structure(Pritchard et al. 2000) Bayesian model-based clustering of genotype data Eigenstrat (Patterson et al. 2006) PCA-based inference of axis of genetic variation
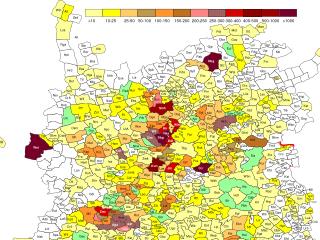
Population structure within Europe (Novembreet al. 2008)
Eigenstrat (Patterson et al. 2006) • Combines Principal Component Analysis and Random Matrix Theory
Eigenstrat (Patterson et al. 2006) • Runtime O(m2n) computation • The challenge: future (current?) genetic data sets n ≥ 500, 000 m ≥ 20, 000 (e.g. WTCCC Nature 2007: 17,000 individuals, 500K snp array) • Can we extend Eigenstrat to this data to be run on a standard desktop? • Assume low rank, k << min(m,n) • Approx algorithm in O(kmn)computation
Randomized PCA Basic steps: • Random projection (approx. preserves distances) • project data onto low dimensional space • do SVD on Y -- similar to SVD on M • Power method : when spectrum decay is slow
Properties of Randomized PCA • Error bound on the k rank approximation : power iteration drives the leading constant to one exponentially fast as i increases! • Top k eigenvalues and eigenvectors can be well approximated in time O(ikmn) • rapid convergence when close to low rank structure (i=1-3) • slowly decaying singular values require more iterations • Clearly no benefit when ik ≈ m << n
Properties of Randomized PCA • Empical observations • we don’t seem to need power iteration, as random projection good enough (data is low rank) • eigenvalue accuracy estimate can be “sloppy” if emphasis is on subspace estimation, assuming a spectral gap • often we care mainly about subspace estimation accuracy
Generalized eigdecomposition • (Semi) supervised dimension reduction • add prior information by means of class labels • linear and non-linear variations: (L)SIR(Li et al. 1991, Wu et al. 2010) • (Non-) linear embeddings • Laplacian Eigenmaps(Belkin and Niyogi 2002) • Locality Preserving Projections(He and Niyogi, 2003) • Canonical Correlation Analysis
Empirical results • Wishart Covariance Structure • independent N(0,1) entries for data matrix • The Wellcome Trust Case Control Consortium (Nature 2007) • Crohn’s Disease; 500K SNP array; 5,000 individuals
Subspace distance metric • Exact method -- subspace A, approx. method -- subspace B (consider column spaces) • Construct projection operators • Define distance metric:(Ye and Weiss, 2003)
Wishart covariance Data matrix: independent N(0,1) entries Runtime improvement over exact