Download

1 / 42
420 likes | 617 Views
Mining Patterns from Protein Structures. Wei Wang University of North Carolina at Chapel Hill. Outline. Introduction Motivation Challenges Graph-based Pattern Discovery in Protein Structures Applications Conclusions Future Directions. Lys. Lys. Gly. Gly. Leu. Val. Ala. His.

E N D
Mining Patterns from Protein Structures Wei Wang University of North Carolina at Chapel Hill
Outline • Introduction • Motivation • Challenges • Graph-based Pattern Discovery in Protein Structures • Applications • Conclusions • Future Directions
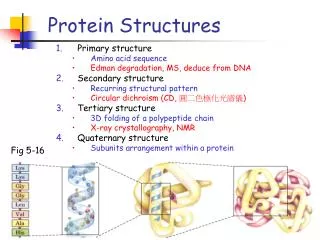
Lys Lys Gly Gly Leu Val Ala His Oxygen Nitrogen Carbon Sulfur Ribbon Introduction • Protein • A sequence from 20 amino acids • Adopts a stable 3D structure that can be measured experimentally
Serine protease active center 1HJ9 1R64 1SSX Introduction • Structure patterns are geometric arrangements of amino acids that are common to a group of different proteins. Three proteins with the same function
Motivations • Structure patterns are useful in: • Protein structure alignment • Protein design • Prediction of protein-protein interactions • Understanding protein folding • Drug design
Goal • Develop techniques to discover structure patterns that are • Efficient • Effective
Growth of Known Structures in Protein Data Bank 35,000 The total number of known protein structures Newly characterized proteins in that year # of structures 1988 2005 Year Challenges • Define mathematical models to represent protein structures • Point set • Labeled graph • Define computational components • Define structure pattern • Specify a matching condition • Design a search procedure • Evaluate the results • computational efficiency and effectiveness
…. The Nature of Protein Structure Data • The ball-stick model is an element-based structure representation • A structure is decomposed into a set of amino acids • Proteingeometry,topology,andattributesare defined with respect to the amino acid set
Components of Pattern Discovery • The definition of patterns • Geometry vs. topology • The matching condition • Measures the fitness of a pattern to a set of protein structures • The search procedure
Related Work Protein Local Structure Comparison Problem Pattern Discovery Pattern Matching • ASSAM, Artymiuk et al., JMB’94 • TESS, Wallace et al., Prot. Sci. ‘97 Sequence-dependent Sequence-independent • TRILOGY, Bradley et al., RECOMB’01 Multi-way comparison Pair-wise comparison • PINTS, Russell, JMB’98 • Geometric Hashing, Fischer et al., Prot. Sci.’94 • Graph Matching, Schmitt et al., JMB’02 • Evolutionary Trace, Lichtarge et al., JMB’96 • FFSM & its variants, Huan et al., ICDM’03, RECOMB’04, CSB’06 Huan et al. Advances in Computers
Our Approach A group of protein structures Represent each structure as a labeled graph Discover frequent occurring subgraphs Map subgraphs to protein structures and obtain structure patterns Predict protein function Identify functional sites in proteins Discover patterns in structure evolution
Outline • Introduction • Graph-based Pattern Discovery in Protein Structures • Labeled graphs and representing structures as labeled graphs • Frequent subgraph mining • Applications • Conclusions • Future Directions
p5 p2 y c b y p1 x a y y d b p4 p3 G1 q1 s1 s4 y b c y y b s2 q2 a a x y y b b s3 q3 G3 G2 Labeled Graphs • A labeled graph is a graph where each node and each edge has a label.
Protein Contact Map • Use a labeled graph to represent a protein structure • Nodesrepresent amino acids,labeled by theidentityof the amino acids • Edgesconnect two amino acids if their Euclidian distance is less than a certain threshold Contact A protein
p5 p2 s1 s4 y y c b b c y y s2 p1 x a a y y y q1 b d b y b s3 p4 p3 q2 G3 G1 a x g2 g3 y y y c b a b g1 q3 G2 G Pattern Matching • A graph G is subgraph isomorphic to a graph G’, denoted by G G’, if • there exists a 1-1 mapping from nodes in G to G’ such that node labels, edges, and edge labels are preserved with the mapping. • A pattern is a graph. Pattern Gmatches G’ if G G’ • Goccurs in G’ if G G’. • With a label set, a graph space is a collection of graphs whose labels are from the set.
Subgraph Mining: Notations Cont. • The support value of a pattern P in a collection of graphs G is the fraction of graphs in G where Poccurs. • Given a collection of graphs G and a threshold 0 < 1, the frequent subgraph mining problem is the identification of all patterns that have support at least .
p5 p2 y c b y p1 x a y y d b p4 p3 G1 y y b c b q1 s1 s4 y b P3 b P2 y b c y y b y + s2 q2 x x a a a a x y y y f=3/3 x f=2/3 b b a f=2/3 b b b b + + P6 P5 s3 q3 + P4 G3 G2 Examples The induced subgraph isomorphism penalizes any unmatched edges = 2/3 b y f=2/3 f=0/3 f=2/3 f = 1/3 f = 3/3 a y b P1 +: induced frequent subgraphs
p5 p2 y c b y p1 x a y y d b p4 p3 G1 b y y y b a c b y q1 s1 s4 b y b P1 b P2 y b c y y b y s2 q2 x x a a a a x y y y f=3/3 x f=2/3 b b a f=2/3 b b b b P6 P5 s3 q3 P4 G3 G2 Examples Maximal frequent subgraph are ones that none of their supergraphs are frequent Other criteria for selecting subgraphs may be incorporated = 2/3 f=2/3 ! P3 !: Maximal frequent subgraphs
Search DAG • Task: identify all frequently occurring subgraphs from a group of graphs, or a graph database • Support anti-monotonicity • Any supergraph of an infrequent subgraph is infrequent • Known as the Apriori property • Level-wise search • Keep all patterns with the same size in memory (poor memory utilization) • Depth-firstsearch • Better memory utilization • May repeatedly search patterns in the DAG (redundant candidates)
Related Work • Level-wise search • AGM: Inokuchi et al., PKDD’00 • FSG: Kuramochi & Karypis, ICDM’01 • Depth-first search • gSpan, Yan & Han, ICDM’02, KDD’03 • FFSM, Huan et al., ICDM’03 • Path-based search • Vanetik, et al., ICDM’02, ICDE’04 • GASTON: Nijssen & Kok, KDD’04 • Tree-based search • SPIN, Huan et al., SIGKDD’04 • Mining with constraints • CSM, Huan et al., CSB’06
The Fast Frequent Subgraph Mining (FFSM) Overview • Graph normalization • Graph Canonical Adjacency Matrix Tree (CAM Tree) • Incremental subgraph isomorphism test Huan et al. ICDM 2003
An arbitrary set Intuitions for Graph Normalization A Graph Space A partial order defined on the graph space A 1-1 mapping A partial order defined on
Graph Normalization • With a partially ordered set (, ),φ: G* → that maps a graph space G* to is a graph normalization function if φ is a 1-1 mapping. • (mapping partial orderφ) Given a graph normalization φandits codomain(, ), we define a binary relation φ G* G* such that P φQ if φ(P) φ(Q) • Claim: φis a partial order
Ideal Normalization • Given a partially ordered codomain (, ),a normalization functionφ: G* → is an ideal normalization if • φinduces a search tree (No redundant candidates) • φ is a subset of the subgraph relation, i.e. for all graphs P and Q, P φQ implies PQ (anti-monotonicity of support )
p’2 P1 P2 P3 P4 P1 P2 P4 P3 P1 P4 P2 P3 b x p’1 y a x a a a x c b x x 0 b c b p’4 p’3 0 x x x y x b b c M1 M3 M2 (P’) y 0 0 x x 0 0 y x b b c p2 p4 x c b x p1 y a x b p3 (P) Graph Canonical Code • The Canonical Code (θ)maps a graph G to a string. • Claim:θ: G* → (*, ) is a graph normalization θ: G* → (*, ) is an ideal graph normalization Code(M1): (1, 1, a)(2, 1, x) (2, 2, b) (3, 1, x) (3, 2, y) (3, 3, b) (4, 2, x) (4, 4, c) Code(M1): (1, 1, a)(2, 1, x) (2, 2, b) (3, 1, x) (3, 2, y) (3, 3, b) (4, 2, x) (4, 4, c) < Code(M2):(1, 1, a)(2, 1, x) (2, 2, b) (3, 2, x) (3, 3, c) (4, 1, x) (4, 2, y) (4, 4, b) < Code(M3): (1, 1, a)(2, 2, c) (3, 1, x) (3, 2, x) (3, 3, b) (4, 1, x) (4, 3, y) (4, 4, b) θ(P) = (1, 1, a)(2, 1, x) (2, 2, b) (3, 1, x) (3, 2, y) (3, 3, b) (4, 2, x) (4, 4, c) • (i, j, Mi,j) (k, l, Mk,l) if • i < k, or • i = k, j < l, or • i =k, j = l, Mi,j Mk,l
FFSM Search • Task: identify all frequently occurring subgraphs from a family of graphs • Depth-firstsearch • Better memory utilization • Apriori property • Eliminate unnecessary isomorphism checks • Graph normalization • Avoid redundant examination • Subgraph isomorphism test is NP-complete • Incremental isomorphism check • Applies to frequent induced subgraph mining with minor modifications +
O = _ _ C C C Performance of FFSM Running time (s) PTE (Predictive Toxicology Evaluation) data set • Contains 340 chemicals • Performances were collected from literatures where experiments were performed with different hardware configurations (400Mhz PIII to 2GHz PIV) • Software downloadable from http://www.cs.unc.edu/~huan • AGM: Inokuchi et al. PKDD’00 • FSG: Kuramochi & Karypis, ICDM’01 • gSpan: Yan & Han, ICDM’02 • FFSM: Huan et al. ICDM’03 • Gaston: Nijssen & Kok, KDD’04
FFSM Scalability Running time (s) Serine protease: • Contains 40 proteins • Contact is defined between every pair of distinct residues if the distance between their C atoms is less than a certain upper-bound (e.g. 6.5 angstrom) • Performances were measured in a single 2GHz PIV CPU with 2GB main memory • gSpanhandles graphs with no more than 254 edges • Gaston runs out of memory
Outline • Introduction • Graph-based Pattern Discovery in Protein Structures • Applications • MotifSpace Architecture • Identify functional sites in proteins • Predict protein function • Conclusions • Future Directions
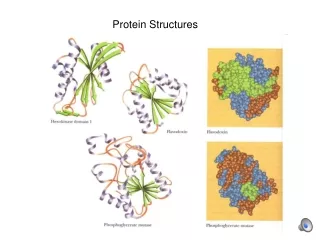
Effectiveness • Serine proteases have three subclasses • Subtilisins • Eukaryotic serine proteases • Prokaryotic serine proteases 1HJ9 1R64 1SSX
Frequent Patterns • 20 highly specific patterns mined from serine proteases # of patterns is the total number of fingerprints a protein has. The coverage of a protein is the fraction of residues which are covered by at least one fingerprint (%), Length (of the protein) is displayed in unit of 200 residues
Patterns’ Biological Relevance 1HJ9 1MD8 1OP0 1OS8 1PQ7 1P57 1SSX 1S83
More Case Studies • Papain-like cysteine proteases • Nuclear receptor ligand binding domains • NADP/FAD binding proteins Papain-like cysteine protease Nuclear Binding domains NADP binding proteins
Predict Protein Function How does a protein function in a biological system? Function Functional motifs carry out protein function 3D structure of a protein
Abr. Name #M #P #M: number of members in a family #P: number of patterns obtained from the family Distinguishing Families with Different Function • TIM barrel Fold contains many proteins with similar structures but different functions Bandyopadhyay, Huan et al. Prot. Sci. ‘06
Functional Inference for 1TWU 1ecs 1twu Yyce SCOP 54598 Antibiotic resistance protein Glyoxalase / bleomycin resistance / dioxygenase superfamily 4 members (SCOP 1.65), 62 family specific spatial motifs unknown function, not in SCOP 1.67, DALI z < 10 in Nov 2004 46 motifs found, structurally similar to the three new non-redundant AR proteins added in SCOP 1.67
G O C A T H S C O P MotifSpace Architecture Biological Experiments Protein Data Bank testable hypotheses Experimental validation protein structures protein family Pattern Filter Pattern Miner Protein Classifier Pattern Validation Subgraph mining Visualization Classification Feature selection structure patterns family-specific patterns Structure Pattern Database Functional Motifs Knowledgebase Indexing & Search Knowledge management Huan et al. ISMB’05 demo, http://escience2-cs.cs.unc.edu/Default.aspx
Summary Goal: pattern discovery in protein structures • Develop labeled graph representations for protein structures • Design algorithms to identify recurring subgraphs in a collection of graphs • Frequent, constrained, maximal, or coherent subgraph mining • Performance evaluation on various data sets • Collaborate with domain experts to evaluate the utility of the algorithms • Predict function for protein structures • Identify structure patterns in protein fold families
Future Work • Pattern discovery in protein structures • Approximate pattern discovery • More applications: • Protein-protein interaction • Protein subcellular localization
Complex Data in Biology Data Models Biological Data Volume
Biological systems at the molecular level Data Analysis in Biological Systems • Challenges: • What are the nature of the data from biological systems? • What are the computational tasks? • How to divide the tasks into a group of computational components? • How to evaluate the results? Source: http://bioinformatics.ca/workshop_pages/bioinformatics/
Acknowledgements • Collaborators: Charlie Carter (UNC School of Medicine), Nikolay Dokholyan (UNC School of Medicine),Leonard McMillan, Jan Prins, Jack Snoeyink,Alexander Tropsha (UNC School of Pharmacy) • Students: Deepak Bandyopadhyay, Yetian Chen (UNC School of Pharmacy), Jun Huan, Jinze Liu, Ruchir Shah (UNC School of Pharmacy), Kiran Sidhu, Xueyi Wang, David Williams, Tao Xie, Jingdan Zhang