Download
1 / 37
400 likes | 438 Views
Explore the transition from 1-D to 3-D band structures in solids, covering real and reciprocal space lattice vectors, Brillouin zones, Fourier transformations, and electronic properties calculations. Dive into the complexity of materials with many orbitals per atom and multiple dimensions. Discover how to plot bands for silicon in various directions within Brillouin zones.

E N D
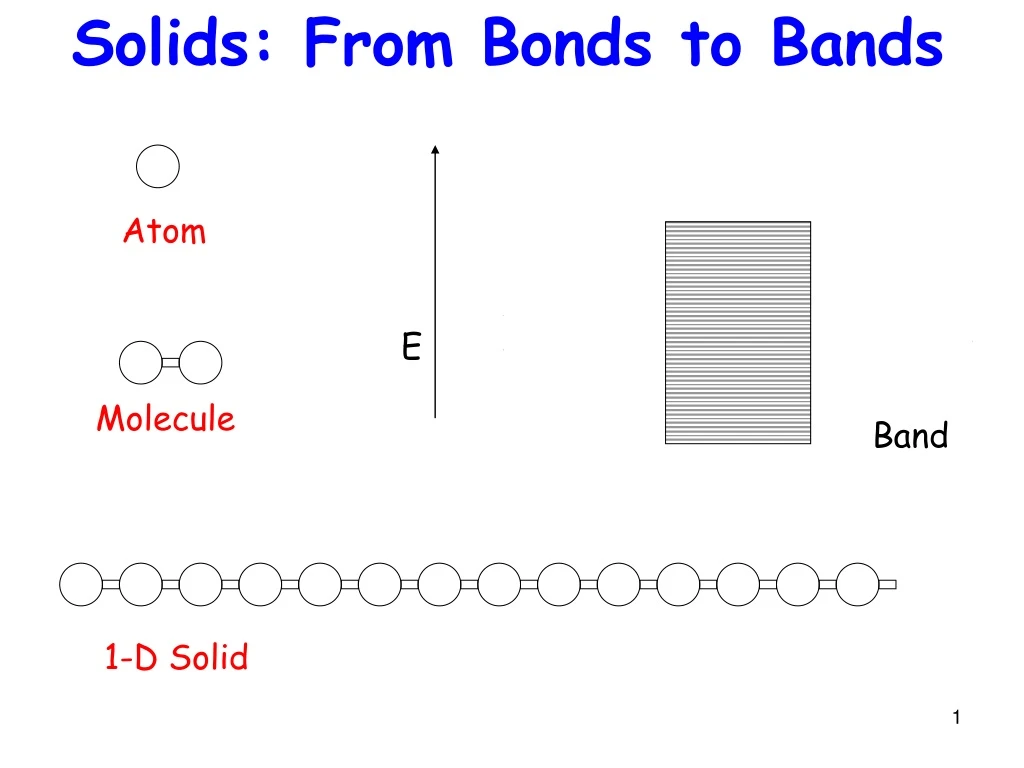
Solids: From Bonds to Bands Levels Bond Molecule Band 1-D Solid Atom E
Real Materials more complex • Many orbitals per atom • Multiple dimensions (3-D) Let us first recap the 1-D bandstructure, so we can see how to generalize it in 3-D. Being systematic helps !!
R = a R = a K x x x x x x -2p/a 0 2p/a Summary of 1D bandstructure STEP 1: Find period in real space STEP 2: Find k-space periodicity (connecting equivalents points in k-space) K.a = 2p STEP 3: Find BZ by bisecting nearest neighbor connectors. This gives the smallest zone in k-space for a non-repeating band. In this case, it’s between –p/a and p/a. On occasion, this may need you to choose a multiatom or multiorbital basis
-p/a p/a 0 n n-1 n+1 Hk = [Hnn] + [Hn,n+1]eika + [Hn,n-1]e-ika Hn,n-1 Hn,n+1 Hnn b bands x x x x x x . . . . . . . . . . . . Summary of 1D bandstructure STEP 4: Choose N allowed k-points by imposing periodic boundary conditions after N unit cells. For complex solids, we may need to choose specific directions. STEP 5: Identify nearest neighbors and find Fourier transform of H terms over this range for each allowed k. Each [Hnn] has size bxb (b: # basis sets) STEP 6: Find eigenvalues E(k). This gives b bands for each k within the BZ. k= (n/N)K, where n=0,1,2,…,N-1, and K=2p/a
Summary of 1D bandstructure STEP 6: Use this bandstructure E-k to calculate DOS D(E), fit parabolas to extract effective mass m*, etc. These are then used for calculating electronic properties like transmission, I-V, etc.
General prescription in 3-D 1 2 4 0 [H]nm 3 Identify real and k-space lattice vectors Identify Brillouin zone Choose grid points along suitable directions in k-space Find H(k) by summing over nearest neighbor H terms with Fourier phases Find eigenvalues to get E-k, which we then use as needed
Finding real-space periodicity (Lattice vectors)
Simple cubic lattice a = (1,0,0)a b = (0,1,0)a c = (0,0,1)a Lattice Vectors Three primitive vectors are ‘coordinates’ in terms of which all lattice coordinates R can be expressed R = ma + nb + pc (m,n,p: integers)
a = a(0, ½, ½) b = a(½, 0, ½) c = a(½, ½, 0) Face-centered cube 6 face center atoms shared by 2 cubes each, 8 corners shared by 8 cubes each, giving a total of 8 x 1/8 + 6 x 1/2 = 4 atoms/cell
a = a(½, ½, ½ ) b = a(-½,-½, ½ ) c = a(½,-½,-½ ) Body-centered cube 8x1/8 corner atom + 1 center atom gives 2 atoms per cell
Finding k-space periodicity (Reciprocal Lattice vectors)
R = ma + nb + pc (m,n,p: integers) Not to be confused with coordinate r of electron which is spread out everywhere K = MK1 + NK2 + PK3 (M,N,P: integers) Need to find reciprocal basis sets K1, K2, K3 such that reciprocal lattice coordinates can be written as First let’s Fourier transform the lattice Coordinates of periodic lattice (ie atoms)
R = ma + nb + pc (m,n,p: integers) To create analogy with 1-D, want K1 to be orthogonal to b and c, and have a 2p overlap with a so that exp(iK.R) = 1 K1 = 2p(b x c)/[a. (b x c)] K = MK1 + NK2 + PK3 (M,N,P: integers) What are lattice vectors in reciprocal space? Pts in k-space spaced by integer # of K’s represent same electronic state
Choosing k values M, N, P unit cells along Reciprocal lattice vectors k = (m/M)K1 + (n/N)K2 + (p/P)K3 m=0,1,2,…(M-1) n=0,1,2,…(N-1) p=0,1,2,…(P-1)
K2 K1 A simple 2-D Example b a In 3-D Reciprocal lattice of BCC FCC !!
Now to find smallest unit cell volume in k-space (Brillouin zone) First let’s do the same in real space (Wigner-Seitz cell)
How to pick a “Primitive” Unit cell? Two atoms in cell. Want to take a smaller ‘primitive’ cell which only contains 1 atom Many ways to do this
How to pick a “Primitive” Unit cell? The special ‘Wigner- Seitz Cell’ is the one that preserves the translational/rotational symmetry of the lattice • Join nearest neighbors • Bisect these lines • Join the bisectors
3-D Wigner-Seitz Cell • Complicated, but it’s the smallest volume in real-space with 1 atom in it • Join nearest neighbors with center atom, draw bisecting planes, and identify volume enclosed by them • To get smallest range of unique k-values for E-k, just need to Fourier transform this! • ie,Wigner-Seitz in Fourier space Brillouin Zone
Choose specific directions within the BZ to plot E-k along
E K along X direction (100, 010, 001 etc) Real Materials more complex • Many orbitals per atom (many bands) • Multiple dimensions (3-D) • Let us look at bands for silicon
E L valley along (111) Real Materials more complex • Many orbitals per atom (many bands) • Multiple dimensions (3-D) • Let us look at bands for silicon L G X
Real Materials more complex • Many orbitals per atom (many bands) • Multiple dimensions (3-D) • Let us look at bands for silicon E E L G X Combine these into 1 figure
Now that we’ve identified k directions,let’s choose a grid of k points, and find the E-k
S S (bx1) m[H]nm{fm} = E{fm} m[H]nmeik.(dm-dn) Try {fm} = {f0}eik.dm (Bloch’s Theorem) Remember these are coeffs of f in atomic {um} basis Solution E{f0} = [h(k)]{f0} where h(k) = n: any unit cell atom, m: all its nearest neighbors Eigenvalues of h(k) E(k) (bandstructure) General prescriptions 1 b basis sets per atom Equation for mth atom 2 4 0 [H]nm 3
Some important materials Cu: Metal
Some important materials GaAs: Direct Bandgap
E DOS Some important materials Si: Indirect Bandgap
3 3 2 2 1 1 2 FCC lattices interpenetrating, displaced by ¼ the body diag Where are the dimers?? Along body diagonal (111 dirn), atoms unequally placed But along x-axis (100) atoms equally placed
With s orbitals alone, dimerization takes care of gap in (111) direction, not (100) • To get gap in (100), need s and p orbitals, since projection of p orbitals unequal • To get indirect bandgap, need s, p, s* orbitals
Approximations to bandstructure Properties important near band tops/bottoms Described through Constant Energy ellipsoids
hh lh Approximations to bandstructure In contrast, valence bands are more warped and are hard to write as parabolas. One uses 6 band k.p for instance..