Download

1 / 26
300 likes | 558 Views
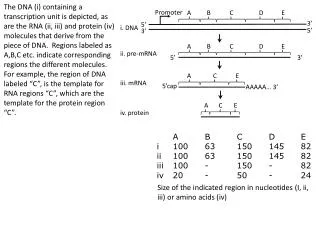
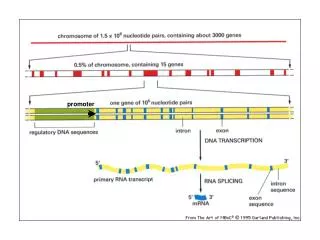
Promoter prediction in eukaryotes. Grupo 5. Eukaryotic Gene Structure. branchpoint site. 5’site. 3’site. exon 1 intron 1 exon 2 intron 2. CAG/NT. AG/GT. Key Eukaryotic Gene Signals. Pol II RNA promoter elements Cap and CCAAT region GC and TATA region

E N D
Eukaryotic Gene Structure branchpoint site 5’site 3’site exon 1 intron 1 exon 2 intron 2 CAG/NT AG/GT
Key Eukaryotic Gene Signals • Pol II RNA promoter elements • Cap and CCAAT region • GC and TATA region • Kozak sequence (Ribosome binding site-RBS) • Splice donor, acceptor and lariat signals • Termination signal • Polyadenylation signal
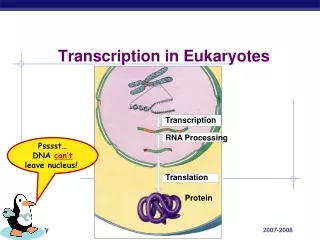
RNA Polymerase creates RNA from DNA • RNA Polymerase I – rRNA • RNA Polymerase II – mRNA • RNA Polymerase III – tRNAs, srRNAs RNA Pol II
Pol II Promoter Elements Exon Intron Exon GC box ~200 bp CCAAT box ~100 bp TATA box ~30 bp Gene Transcription start site (TSS)
Transcriptional regulatory region • TFs play a significant role in differentiation in a number of cell types • The fact that ~ 5% of the genes are predicted to encode transcription factors underscores the importance of transcriptional regulation in gene expression (Tupler et al. 2001 Nature.409:832-833) • The combinatorial nature of transcriptional regulation and practically unlimited number of cellular conditions significantly complicate the experimental identification of TF binding sites on a genome scale • Understanding the transcriptional regulation is a major challenge • Computational approaches to identify potential regulatory elements and modules, and derive new, biologically relevant and testable hypothesis
Pol II Promoter Elements • Cap Region/Signal • n C A G T n G • TATA box (~ 25 bp upstream) • T A T A A A n G C C C • CCAAT box (~100 bp upstream) • T A G C C A A T G • GC box (~200 bp upstream) • A T A G G C G nGA
Pol II Promoter Elements TATA box is found in ~70% of promoters
Finding Eukaryotic Genes Computationally • Content-based Methods • GC content, hexamer repeats, composition statistics, codon frequencies • Site-based Methods • donor sites, acceptor sites, promoter sites, start/stop codons, polyA signals, lengths • Comparative Methods • sequence homology, EST searches • Combined Methods
Methods • Neural nw trained on TATA and Inr Sites allowing a variable spacing between sites. NN-GA approach to identify conserved patterns in RNA PolII promoters and conserved spacing among them (PROMOTER2.0). • TATA box recognition using weight matrix and density analysis of TF sites. • Usage of linear (TSSD and TSSW) /quadratic (CorePromoter) discriminant function. The function is based on: • TATA box score • Base-pair frequencies around TSS (triplet) • Frequencies in consecutive 100-bp upstream regions • TF binding site prediction • Searches of weight matrices for different organism against a test sequence (TFSearch/ TESS). MatInspector and ConInspector allows user-provided limits on type of weight matrix, generation of new matrices etc. • Testing for presence of clustered groups (or modules) of TF binding sites which are characteristics of a given pattern of gene regulation.
Promoter Databases: • TRANSFACis a database on eukaryotic cis-acting regulatory DNA elements and trans-acting factors. It covers the whole range from yeast to human. • Biological Databases/BiologischeDatenbanken GmbH • In release 4.0, it contains 8415 entries, 4504 of them referring to sites within 1078 eukaryotic genes, the species of which ranging from yeast to human. Additionally, this table comprises 3494 artificial • sequences which resulted from mutagenesis studies, in vitro selection Procedures starting from random oligonucleotide mixtures or from specific theoretical considerations. And finally, there are 417 entries with consensus binding sequences given in the IUPAC code. • MatInspectorSearch for potential transcription factor binding sites in your own sequences with the matrix search program MatInspector using the TRANSFAC 4.0 matrices. • FastM A program for the generation of models for regulatory regions in DNA sequences. FastM using the TRANSFAC 3.4 matrices. • PatSearchSearch for potential transcription factor binding sites in your own sequences with the pattern search program using TRANSFAC 3.5 TRRD 3.5 sites. • FunSitePRun interactively FunSiteP. Recognition and classification of eukaryotic promoters by searching transcription factor binding sites using a collection of Transcription factor consensi. http://www.gene-regulation.com/
TESSTranscription Element Search System Computational Biology and Informatics Laboratory, School of Medicine, University of Pennsylvania, 1997 http://www.cbil.upenn.edu/cgi-bin/tess/tess33?WELCOME Eukaryotic Promoter Database Swiss Institute for Experimental Cancer Research • The Eukaryotic Promoter Database is an annotated non-redundant collection of eukaryotic POL II promoters, for which the transcription start site has been determined experimentally. • The annotation part of an entry includes description of the initiation site mapping data, cross-references to other databases, and bibliographic references. • EPD is structured in a way that facilitates dynamic extraction of biologically meaningful promoter subsets for comparative sequence analysis. • EPDEX is a complementary database which allows users to view available gene expression data for human EPD promoters. • EPDEX is also accessible from the ISREC-TRADAT database entry server. http://www.epd.isb-sib.ch/ Prediction of transcription factor binding sites by constructing matrices on the fly from TRANSFAC 4.0 sites. AliBaba http://darwin.nmsu.edu/~molb470/fall2003/Projects/solorz/ http://www.epd.isb-sib.ch/TRADAT.html
Neural Networks (PROMOTER 2.0) http://www.cbs.dtu.dk/services/promoter/ Density of TF from EPD (PromoterScan) http://bimas.dcrt.nih.gov/molbio/proscan/ Searches of weight matrices against a test sequence (TFSearch/TESS) http://www.cbil.upenn.edu/cgi-bin/tess/tess
Tools • ORF detectors • NCBI: http://www.ncbi.nih.gov/gorf/gorf.html • Promoter predictors • CSHL: http://rulai.cshl.org/software/index1.htm • BDGP: fruitfly.org/seq_tools/promoter.html • ICG: TATA-Box predictor • PolyA signal predictors • CSHL: argon.cshl.org/tabaska/polyadq_form.html • Splice site predictors • BDGP: http://www.fruitfly.org/seq_tools/splice.html • Start-/stop-codon identifiers • DNALC: Translator/ORF-Finder • BCM: Searchlauncher
Problems arising in gene prediction. • Distinguishing pseudogenes (not working former genes) from genes. • Exon/intron structure in eukaryotes, exon flanking regions – not very well conserved. • Exon can be shuffled alternatively – alternative splicing. • Genes can overlap each other and occur on different strands of DNA. • Only 2% of human genome is coding regions • Intron-exon structure of genes • Large introns (average 3365 bp ) • Small exons (average 145 bp) • Long genes (average 27 kb)
Outstanding Issues • Most Gene finders don’t handle UTRs (untranslated regions) • ~40% of human genes have non-coding 1st exons (UTRs) • Most gene finders don’t’ handle alternative splicing • Most gene finders don’t handle overlapping or nested genes • Most can’t find non-protein genes (tRNAs)
Gene expression is also influenced by the region upstream of the core promoter and other enhancer sites. • Eukaryotic sequences show variation not only b/w species but also among genes within a species. Hence, a set of promoters in an organism that share a common regulatory response is analyzed • The programs can predict 13-54% of the TSSs correctly, but also each program predicted a number of false-positive TSSs.
Bottom Line... • Gene finding in eukaryotes is not yet a “solved” problem • Accuracy of the best methods approaches 80% at the exon level (90% at the nucleotide level) in coding-rich regions (much lower for whole genomes) • Gene predictions should always be verified by other means (cDNA sequencing, BLAST search, Mass spec.)