Download
1 / 39
390 likes | 450 Views
Learn about the microscopic mechanisms and magnetic structure determination in magnetic molecular compounds using neutron diffraction techniques.

E N D
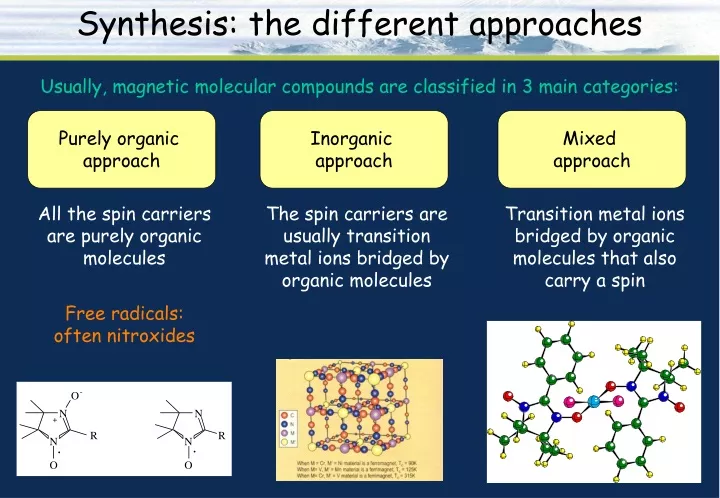
Synthesis: the different approaches Usually, magnetic molecular compounds are classified in 3 main categories: Purely organic approach Inorganic approach Mixed approach All the spin carriers are purely organic molecules The spin carriers are usually transition metal ions bridged by organic molecules Transition metal ions bridged by organic molecules that also carry a spin Free radicals: often nitroxides
The magnetization distribution 4Question ? What are the microscopic mechanisms at the origin of the different magnetic behaviors in these molecules ? 4Main problem: complexity of these systems ! The limits of many models used for “classical” compounds are reached, and reliable theoretical frameworks have still to be built up 4A crucial piece of information: the magnetization distribution Where are localized unpaired electrons in the molecules (magnetic orbitals) ? • TPV free radical : one unpaired electron Where ? In the SOMO, wich is built from individual atomic orbitals -> the magnetic moments are strongly delocalized over the molecules Shape of the SOMO ? N C Neutron Diffraction ! H
Neutron diffraction Neutron: two main interactions in condensed matter • Nuclear interaction: with the nuclei of atoms --> crystallography some advantages compared to x-ray : • low temperatures routinely • precise localisation of H atoms • Magnetic interaction : between the magnetic moment of the neutron and the magnetic field H created by unpaired electrons --> most widespread use of neutron diffraction: magnetic structure determination, i.e. determination of the direction and the amplitudes of magnetic moments in the ordered state.
Neutron diffraction Magnetic structure determination: in this kind of studies, it is assumed • the magnetic moment on each atom is a vector • the magnetization distribution around each atom comes from atomic calculations for free ions -> ‘form factors ’ tabulated in the International Tables of crystallography What is the form factor of such a molecule ? To go beyond this approximation -> to investigate these distributions on a sub-atomic scale: Polarised neutron diffraction Classical polarised beam technique
Classical polarized beam technique 4Q = scattering vector 4P0 = polarisation of the incident neutron beam 4FN = nuclear structure factor Fourier component of the nuclear density 4M = magnetic interaction vector proportional to the Fourier component of the magnetization distribution
Increased Sensitivity 4Suppose M = 0.1 x FN (10%) 4Unpolarized beam: I = FN2 + (0.1 x FN)2 = 1.01 FN2(1% effect on the intensity) 4Polarized beam: I+ = FN2 + (0.1 x FN)2 + 2 FN x (0.1 x FN) = 1.21 FN2(20% effect) I- = FN2 + (0.1 x FN)2 - 2 FN x (0.1 x FN) = 0.81 FN2(20% effect)
Limitations • Interference: -> the magnetic and nuclear signal should appear at the same positions in reciprocal space -> the magnetic cell and the nuclear cell should have the same periodicity -> Limitations: in the different classes of compounds that could be studied by this technique • Paramagnets under an applied field (to induce a long range magnetization) • Saturated ferromagnets (single domain) • Ferrimagnets • Some antiferromagnets (a few particular cases, with several Bravais sublattices and in-phase nuclear and magnetic structure factors )
Experimental conditions 4Experiments performed on a single crystal, usually in the paramagnetic state 4 In an applied magnetic field to induce a long range order of the magnetization 4 One measures the ratio R (“flipping ratio”) between the scattered intensities for P0 and -P0 4 From the measurement of R and the knowledge of FN(Q), one deduces the magnetic structure factors FM(Q) of the different Bragg reflections Q with both sign and amplitude (centric cases) +Fourier components of the magnetization distribution 4 They contain information on : +all the atoms carrying magnetization +all the shells involved +spin + orbital contributions (if present) M(r) = Ms(r) + M(r)
Data treatment • The experiment gives the magnetic structure factors FM(Q) of the different Bragg reflections Q with both sign and amplitude (centric cases) •The FM(Q)’s are the Fourier components of the magnetization distribution M(r) • To retrieve the magnetization distribution in real space,an inverse Fourier problem should be solved Several methods: • Direct (model free) methods +Model refinements • use nothing but the experimental data • necessary step before any attempt to refine a model + Raw Fourier transform + Maximum of Entropy analysis • they require the system to be well enough understood for a model to be proposed + Multipolar expansion + Wave function analysis
Magnetic Molecular Clusters Molecules formed by a large number of strongly interacting metal ions +Used as models of nanometric-size single-domain magnetic particles +They offer the opportunity to study the magnetism at a mesoscopic scale using macroscopic samples (without broad distributions in size and shape) +Clusters with high spins in the ground state and large Ising-type anisotropy have attracted a lot of interest in recent years. • Superparamagnetic behaviour • Slow relaxation of the magnetization • Quantum tunneling of the magnetization +Large interest to study the exact nature of the ground state of these clusters (coupling scheme) to explain the observed properties.
The Fe8 Cluster Cluster formed of 8 Fe3+ ions (s = 5/2) Competing antiferromagnetic interactions -> S = 10 The 8 iron atoms are splitted in two sets: • 6 iron atoms with moments parallel to the applied field • 2 iron atoms with moments antiparallel to the applied field J. Am. Chem. Soc. 121 (1999) 5342
The Mn10 Cluster • Cluster formed of 6 Mn2+ ions (Mn1 and Mn2, S=5/2) and 4 Mn3+ ions (Mn3, S=2) Antiferromagnetic interactions -> S = 12 • Magnetization only at the Mn sites (nothing on the ligands) • Mn1 opposed to both Mn2 and Mn3 • Spherical shape on Mn1 and Mn2 (S=5/2) • Elongated on Mn3(S=2) Physica B 241-243 (1998) 600
The free radical TPV N C H • Stable free radical • Spin 1/2 • Zig-zag chains in the crystal • Antiferromagnetic ordering TN = 1.8 K • delocalization on the phenyls • sign alternation on the phenyls • negative density on the C • inequivalent nitrogen atoms: N1, N5 : 0.54(1) mB N2, N4 : 0.15(1) mB
Isolated Free Radicals • Spin delocalisation the magnetism of a free radical is attributed to an unpaired electron in the SOMO, built from individual atomic orbitals.The magnetic moments are no longer well localized around one atom, but delocalized over the molecules • Spin polarisation Concern the sign of the magnetization distribution. In a neutron experiment, the external applied field aligns the spin density Negative spin density ? Trivial case: several spin carriers, with an antiferromagnetic coupling -> regions of positive and negative spin densities Free radicals: more subtle because only one unpaired electron -> Intramolecular exchange interaction (positive spins attracted by the positive spin on the SOMO, leaving the negative spins on the nodes of the SOMO. • Shape of the distribution: 2p character, bonding, antibonding …
Interacting Radicals What are the modifications on the magnetization distribution due to the magnetic interactions in the crystal ? • hydrogen bonds • through atoms of the substituents • direct coordination with a metal center
The free radical tempone O N H C Changing R -> flexible chemical structures -> fine “tuning” of magnetic interactions
The free radical tempone O N H C
The free radical tempol O O N N H C O Tempol Tempone H
The free radical tempol O N Tempol O H
Phenyl Nitronyl Nitroxyde H C O Phenyl derivative 4 Curie law down to very low temperatures 4 Very little interactions with neighboring molecules +Well isolated radical The knowledge of the ground state of this isolated radical necessary to understand the properties of the interacting derivatives N Phenyl derivative
Phenyl Nitronyl Nitroxyde Maximum of entropy projections of the magnetization distribution : model free, no assumption on the shape
Phenyl Nitronyl Nitroxyde J. Am. Chem. Soc. 116 (1994) 2019
The free radical NitPy(C=C-H) J. Am. Chem. Soc. 122 (2000) 1298
Copper Phenyl Nitronyl Nitroxide Complex N O Cu O N J. Am. Chem. Soc. 115 (1993) 3610
Experiment vs ab-initio calculations Polarized neutron experiment: roughly 2 weeks of beam time in a cryomagnet + It is necessary to know very well the low temperature crystal structure --> another weak on a 4 circle do we have to measure or calculations are reliable enough ?
Ab-initio Calculations 4Problem ! Because of the electron-electron repulsion term, no analytical solution+ approximations 4Two main families of ab-initio calculations • Methods based on the Hartree-Fock approximation • Methods based on the local density approximation
Hartree-Fock approximation • The molecular wave function is expressed as a Slater determinant of single particle atomic wave functions • The hamiltonian is replaced by a sum of single particle hamiltonians, including kinetic energy, Coulomb attraction by the nuclei and the Coulomb repulsion by the other electrons + this last term is calculated assuming that the other electrons are distributed over their wave function -> self consistent calculations 4Problem : exchange - correlation ! Electrons don’t stay unperturbed on their wave function when another electron is in the neighborhood -> exchange correlation hole 4How to take it into account ? • polarised basis sets (d and f orbitals for 2p electrons) • perturbation theory -> admixing of excited states according to the level of perturbation : MP2, MP3 (Moller-Plesset), or full CI (configuration interaction) -> more and more time consuming 4Restricted Hartree-Fock: each molecular orbital is doubly occupied (the dsp is > 0) 4Unrestricted Hartree-Fock: the orbitals for spin up and spin down are different (the dsp may be negative)
Local density approximation (LDA) 4Hohenberg - Kohn theorem • The energy of an ensemble of electrons is a functional of the charge density and the ground state minimizes this functional • This functional contains a kinematic term, a Coulomb term and an exchange-correlation term 4Problem Nobody knows the analytical expression of the exchange-correlation term ! 4Approximation • The exchange-correlation term for an electron in a crystal is taken as the one for an homogeneous interacting electron gas in a box ! • This approximation has been extended to magnetic systems by introducing a functional for spin up electrons and one for spin down electrons (Local spin density approximation, or LSD) 4To go beyond: corrections of the exchange-correlation term : non local potential, gradient methods ....
The free radical tempone Delocalization:calculated by both method much smaller than observed experimentaly LSD: stronger delocalization (carbons) than Hartree - Fock
Phenyl Nitronyl Nitroxyde J. Am. Chem. Soc. 116 (1994) 2019
The free radical tempol Both calculation agree with a transfer from O to N compared to the tempone Influence of the H bondstronger for Hartree-Fock Population on H: calculated one order of magnitude too small compared to the experiment
Copper Phenyl Nitronyl Nitroxide Complex LSD : 0.58 mB on Nit, -0.19 on Cu, far from the 2/3 -1/3 2/3 observed experimentaly
Conclusion Ab-initio calculations : You can’t always get what you want ..... But if you try sometimes, you might find You get what you need ... (Sir Michael Philip Jagger & Keith Richards) Experiments : Still required !!