Download

1 / 87
870 likes | 1.08k Views
Genome Sequence determination. 陳中庸. E-mail: cychen@cycu.edu.tw Web site: www.cychen.idv.tw. Complete Microbial Genomes. Genome what now?. Sequencing is … Determining the full nucleotide sequence of one strain of an organism

E N D
Genome Sequence determination 陳中庸 E-mail: cychen@cycu.edu.tw Web site: www.cychen.idv.tw
Genome what now? • Sequencing is… • Determining the full nucleotide sequence of one strain of an organism • Making predictions of genes within that sequence & predicting the function of those genes • HARD!!!! • Sequencing requires… • Time • Money • People • Computers
Genome what now? • Before Sequencing … • Nature of an organism • Genetic code • Genome size • Genome structure • Sequencing means… - Bioinformatic - Functional Assay - More….
Organism Selection Library Creation
Organism Selection Library Creation Sequencing
Organism Selection Library Creation Sequencing Assembly
Organism Selection Library Creation Sequencing Assembly
Organism Selection Library Creation Sequencing Assembly
Organism Selection Library Creation Sequencing Assembly Gap Closure
Organism Selection Library Creation Sequencing Assembly Gap Closure
Organism Selection Library Creation Sequencing Assembly Gap Closure Finishing
Organism Selection Library Creation Sequencing Assembly Gap Closure Finishing Annotation
Organism Selection Library Creation Sequencing Assembly Gap Closure Finishing Which steps are computationally expensive? Annotation
Organism Selection Library Creation Sequencing Assembly Gap Closure Finishing Annotation
Organism Selection Library Creation Sequencing Assembly Gap Closure Finishing Which steps have not already been exceptionally well studied? Annotation
Organism Selection Library Creation Sequencing Assembly Gap Closure Finishing Annotation
Organism Selection Library Creation Sequencing Assembly Gap Closure Finishing Which step has not been subjected to a variety of approaches? Annotation
Organism Selection Library Creation Sequencing Assembly Gap Closure Finishing Annotation
Organism Selection • Nature of an organism: Pathogen? • Genetic code • Genome size • Genome structure
Vibrio vulnificus Strain: YJ016 Genome Size: 5.2 Mb Source: Southern Taiwan Significance: Virulence Strategy: Whole Genome Shotgun Sequencing Coverage: 10X
Organism Selection • Nature of an organism: Pathogen? • Genetic code: Special Code? • Genome size • Genome structure
Genetic Code Tables http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi?mode=c
Organism Selection • Nature of an organism: Pathogen? • Genetic code: Special Code? • Genome size: How many Megabases? • Genome structure
Organism Selection • Nature of an organism: Pathogen? • Genetic code: Special Code? • Genome size: How many Megabases? • Genome structure: Linear/Circular Chromosome? How many?
How to sequence a complete genome? Sizes of bacterial genomes vary between :Mycoplasma genitalium and Myxobacteria: 0.6 Mb to ~13 Mb • reading length of DNA sequencing reactions is just ~600 bp (= 0.0006 Mb) ⇒ a subdivision of the genome is obviously necessary • If the genome needs to be subdivided into small pieces of suitable sizes for sequencing, then • Individual sequences/fragments need to be ordered somehow into their "native" order • Therefore, overlaps between each other are necessary in order to re-assemble the pieces ⇒ there are two main sequencing strategies: 1. whole genome shotgun sequencing 2. ordered shotgun sequencing
Two ends are overlapped • Non overlapped • Plasmid percentage in contigs
Library Creation • Team Works • QC control • Time Table • Budget • Paper
Standard Operation Procedures of a Genome project A. Decision Mapping Protocol 1 QC PCR Confirm Protocol 2 B. Library Protocol 3 DNA purification Protocol 4 PFG QC FISH 決定盤數 PCR confirm Protocol 5 Shotgun Library Picking Print Labels C. Sequencing QC Protocol 6 Plasmid DNA Sequencing Reactions Dye Primers Protocol 7 QC Dye Terminator Protocol 8 Gel Running Protocol 9 377 QC Protocol 10 3700 D. Finish Protocol 11 Assemble Protocol 12 Annotation
Library (1) Random Shearing of Genomic DNA • Restriction enzyme: • Sau3AI (GATC)--- affected by CG methylase • MboI (GATC) – affected by dam methylase • -- not affected by CG methylase • 2. Sonication: • Sonication – Bal31 repair – T4 DNApolymerase – Sizing – • Recover –Ligation • 3. GeneMachine: easy sizing by filter
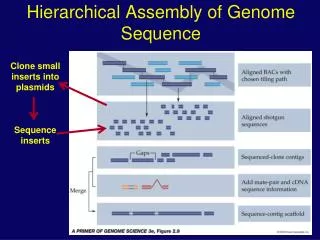
Library (2) Library clones & Sequencing clones Chromosome I Chromosome II 3.3 Mb 1.8 Mb Shotgun library Library 1: 2.5-3.5 kb inserts 7X Coverage Library 2: 5.5-7.5 kb inserts 3X Coverage Library 3: 30 kb inserts Cosmid library 10X Clone Coverage, 0.4X Sequence Coverage Sequenced for both ends Sequenced for both ends Sequenced for both ends Assemble the reads by using phred/phrap/consed softwares Contig 1 Contig 2 Contig 3 Closing the gaps by primer walking, PCR or re-sequencing Annotation
Library (2) Library clones & Sequencing clones 5,000,000 bp 1000 bp/per clone 5,000,000/1000 = 5000 clones =52 x 96 well plates 10 x redundancy 52 x10 x 96 wells plates Library clones Both ends sequencing 2 x 52 x 10 x 96 well plates ≒ 1000 plates Sequencing clones
Sequencing (1) Time table • 377:2 runs/per day (one run for one 96 well plate) • 3700 : 6 runs/per day (POP6) • 8 runs/per day (POP5) • 3730 : 12 runs/per day 2. 377 x 2 sets = 4 runs/per day 3700 x 2 sets = 6 x1 + 8 x 1 = 14 runs/per day total 18 runs per day 3. 1000 plates / 18 = 56 days = 11 weeks (3 months) 4. Today, 3730 for 4 sets = 48 runs/per day; 1000 plats /48 = 20 days
Sequencing (2) Cost
硬體設施 ABI 377 ABI 3700 MegaBace 4000 ABI 3730XL
The automated production line for sample preparation at the Whitehead Institute, Center for Genome Research. The system consists of custom-designed factory-style conveyor belt robots that perform all functions from purifying DNA from bacterial cultures through setting up and purifying sequencing reactions.
5X coverage 359 328 279 Assembled contigs 245 243 166 Assembled reads Reads vs. Assembled Contigs
5X coverage Assembled size (Mbps) 5.17 5.12 5.13 5.10 5.08 5.07 Assembled reads Reads and Assembled Size
What is Gap Closure? • What are gaps? • Unsequenced regions located between assembly generated fragments of contiguous sequence (contigs) • What causes gaps? • Host toxicity, secondary structure, ??? • Back to “gap closure” • Producing, purifying, and sequencing, or locating, the missing regions of DNA
How Can I Close Gaps? • Genome Walking • Blind PCR extension of contigs • Multiplex PCR • Combinatorial trial of every contig pair • Read Pair Analysis • Use information stored by the assembler to suggest alignments, then PCR • Comparative Alignment
Comparative Alignment(the Bioinformatics Approach) • Find locations where contigs are homologous to known sequences • Determine if any contigs share homology in the same region of the same sequence • Design primers • Conduct PCR with those primers • Sequence that product and use that sequence to close the gap
Blast Organism X(cross) - Comparison • Compares contig ends to NCBI “nr” database with BlastN • Parses all hits and finds biologically possible contig pairs • Using the flanking sequence and Primer3, designs primers that will produce a PCR product spanning that gap