Download

1 / 38
390 likes | 632 Views
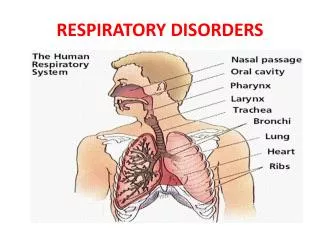
Inherited Respiratory System Disorders. Cystic Fibrosis (CF). CF is the most common life-limiting autosomal recessive disorder in white population. CF is chronic , progressive, and frequently fatal genetic disease of the mucous glands .

E N D
Cystic Fibrosis (CF) CF is the most common life-limiting autosomal recessive disorder in white population. CF is chronic, progressive, and frequently fatal genetic disease of the mucous glands. Affects the respiratory and digestive systems in children and young adults. An average person has a lifespan of 40 years with the right treatment.
Cystic Fibrosis • Cystic Fibrosis is caused by a defective CFTR gene which codes for a Na+ and Cl- transporter found on the surface of epithelial cells of lungs and other organs. • CFTR - Cystic Fibrosis Transmembrane conduction Regulator
Cystic Fibrosis • Autosomal recessive • Gene cloned in 1989: “CFTR” • 1601 mutations in CFTR known to cause CF • An extensive amount of information is known about CFTR Science, September 1989
CFTR Mutation Epithelial cell dysfunction Disease manifestations Lungs Sinuses Pancreas Liver Biliary duct Bones Vas deferens Pathophysiology of CF ?
CF Lung Chloride does not get into airway; more sodium leaves; mucus is thick
Airway Cross Sectional View Mucus layer Pericellular layer with cilia Epithelial cell layer Knowles & Boucher 2002;109:571
Required Geometry for Effective Mucociliary Clearance Knowles & Boucher 2002;109:571
Consequences of CFTR Deficiency on Airway Clearance Knowles & Boucher 2002;109:571
CF Gene Mutation Ion Transport Abnormalities Altered Airway Environment Infection Inflammation Tissue Damage Pathophysiology of CF Lung Disease R. Simon
Pathophysiology of CFLung Disease CF Gene Mutation Recurrent Bronchitis Bronchiectasis Chronic Respiratory Failure Source Undetermined Death R. Simons
Clinical Manifestations Respiratory Tract • Cough is the most constant symptom. • Wheezing • Recurrent chest infection • Cyanosis is a late sign • Atelactasis, hemoptysis, pneumothorax, and corpulmonale • Sinusitis, nasal polyps
Clinical Manifestations…. Intestinal Tract • Meconium ileus 10-20% • Meconium plug syndrome (meconium ileus equivalent)…more than 85% of patients showed evidence of maldigestion from exocrine pancreatic insufficiency. 3. Bile or acid reflux with oesophagitis 4. Sub acute appendicitis 5. Rectal prolapse 6. Failure to thrive 7. Fat-soluble vitamin deficiency manifestation.
Clinical Manifestations… Biliary Tract • Biliary cirrhosis symptomatic in 2-3% . • Ascitis, Jaundice, hematemesis, esophageal varices • Neonatal hepatitis
Clinical Manifestations… • Diabetes Mellitus… 8% after the age of 10. • 95% of males are azoospermic because of failure of development of wolffian duct structure. • Secondary amenorrhea • Cervicitis • Hypochloremic alkalosis
Diagnostic criteria for CF At least one of the following: 1) One or more clinical manifestations of CF • Meconium ileus • Chronic bronchitis / bronchiectasis • Chronic infection of the paranasal sinuses • Pancreatic insufficiency • Salt loss syndromes • Male infertility due to congenital bilateral absence of the vas deferens 2)History of CF in a sibling
Diagnostic Criteria for CF At least one of the following: 1)Elevated sweat chloride test • Identification of a mutation in each CFTR gene known to cause CF
The Sweat Test “Gold Standard” for testing over 40 years - painless - inexpensive - gives definite answers Results; • Cl- ≥ 60 mEq/L Positive • 40-60 mEq/L ?
G551D N1303K W1282X 621+1G T R553X 1717-1G A R1162X R117H Δ I507 3849+10kbC T R347P Genotyping for CF Diagnosis Population Frequency of Specific CFTR Mutations Causing CF • 1601 CFTR mutations known to cause CF • Only 25 mutations have a frequency > 0.1% ΔF508 G542X 0 10 20 30 40 50 60 70 Frequency, % CF Genetic Analysis Consortium
Genotyping for CF Diagnosis • Current commercial screening tests • Look for presence of between 25 - 100 mutations • These will detect a CF allele only ~90% of time • For a group of patients with known CF, genotyping would be diagnostic in only ~81% of patients • Screening for most common mutations is not as sensitive as sweat testing (98%) to diagnose classic CF
Genetic Diagnosis of CF • Tests becoming commercially available for detecting mutations more broadly • PCR used to amplify all exons and surrounding splice sites • Heteroduplex formation screening and/or sequencing • Analysis for large deletions and duplications • Cost ~ $2,500
CF: A Disease for aMultidisciplinary Team The Principles of management: • To Allow the child and his family, as far as possible, to enjoy a normal lifestyle. • To minimize the emotional problems that invariably develop. • To prevent, or at least retard as far as possible, progressive lung disease. • To achieve optimal nutrition and maintain normal growth
CF Treatment • PHYSIOTHERAPY and physical activity • ANTIBIOTIC THERAPY • BRONCHODILATOR THERAPY • Beta2 agonist • ANT-INFLAMMATORY AGENTS • Corticosteroids for Bronchopulmopnaryaspergillosis • TREATMENT OF PULMONARY COMPLICATIONS
Hereditary Pulmonary Emphysema“alpha1 antitrypsin deficiency” • Emphysema - abnormal permanent enlargement of the airspace distal to the terminal bronchioles, accompanied by destruction of their walls and without obvious fibrosis. • Hereditary deficiency of α1-antitrypsin inhibitor accounts for about 2% of emphysema cases. • Alpha1 antitrypsin deficiency (AATD) is a autosomal recessive disorder characterized by a predisposition to emphysema and cirrhosis.
Alpha1 antitrypsin is a serin proteinase inhibitor that protect connective tissue of the lungs from the elastase released by leucocytes. Liver damage arises not from the deficiency of the protease inhibitor, but from pathological polymerization of the variant alpha1-antitrypsin before its secretion from hepatocytes.
More than 75 alleles of the α1-antitrypsin inhibitor gene have been described. The three main phenotypes are ; MM (normal), MZ (heterozygous deficiency) and ZZ(homozygous deficiency).
AATD is a Protein Folding Disease • Protein folding is the process by which an unfolded polypeptide chain folds in to a specific native and functional structure • Defective protein folding is an important mechanism underlying the pathogenesis of many diseases
Abnormal Folding and Polymerization of AAT • The most common and severe form of AAT deficiency is caused by e Z mutation, a single base substitution (Glu-342-lys) in the AAT gene. • This slows the rate of protein folding in the cell • Allowing the accumulation of an intermediate which polymerizes Impeeding its release • Leading to plasma deficiency AAT Polymer
Pathogenesis of Lung Damage in AATD AAT Clinical Case
Lung Related Clinical Manifestations • Emphysema • Presenting Symptoms: • Dyspnea (most common symptom) • Cough, phlegm production and wheezing • Bronchodilator responsiveness • Differences with patients w usual COPD • Earlier Age • Bullous changes prominent in lung bases • > 90 % of ZZ phenotype have lung bases involved • Limited to lung bases in 24 %Found exclusively in • Asthma and Bronchiectasis: • Relationship not proven
Diagnosis • Measure AAT level • Mutation analysis
Asthma • Asthma is a disorder that causes the airways of the lungs to swell and narrow, leading to wheezing, shortness of breath, chest tightness, and coughing. • Asthma is caused by inflammation in the airways. Interest in finding a genetic susceptibility locus is based on the heritability that asthma shows. • Children with one asthmatic parent 3-6 times more likely to develop asthma than a child with two normal parents1. • Children with two asthmatic parents 10 times more likely to develop asthma than normal1.
Asthma: a complex phenotype « Not a single disease entity but made up of various overlapping phenotypes … in people with different genetic predisposition & susceptible to different environmental triggers » OR « A symptom (as fever): the clinical manifestation of several distinct diseases Wenzel, Lancet, 2006
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y Strategies used to identify genes involved in asthma-related phenotypes No Hypothesis Genome-wide screen approach Linkage studies ~ 400 genetic markers (microsatellites) Genome-wide association studies ~ 300 000 genetic markers (SNP) Fine mapping Associations Gene discovery Hypothesis-driven Biological studies Candidate gene approach
> 20 genome screens conducted to date • Populations: • Europeans+++, Australians, North-Americans, Chinese, Japanese Regions most often replicated across populations Phenotype linked to several regions: polygenic? One region linked to several phenotypes: one pleiotropy gene or several genes in the same region?
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y FEV1 SPTQ 21q21 SPT 17q22 IgE 12p13 FEV1 6q14 MultiRAST FEV1 IgE Asthma EOS SPTQ SPT BR GENOME SCANOF 295EGEAFAMILIES for 8 asthma-related phenotypes Bouzigon et al, Hum Mol Genet 2004
Candidate gene approach > 500 association studies of asthma phenotypes (Ober & Hoffjan 2006) 118 genes associated to asthma or atopy phenotypes 54 genes found in 2 to 5 independent studies 15 genes found in 6 to 10 independent studies 10 genes found in > 10 independent studies IL4, IL13, CD14, IL4RA, ADRB2, HLA-DRB1, HLA-DQB1, TNF, FCER1B, (ADAM33)
Pulmonary Fibrosis associated with known genetic causes • Neuofibromatosis • HermenskyPuldak Syndrome • Gaucher Disease • Niemann-Pick Disease • Tuberous sclerosis