Download

1 / 23
270 likes | 354 Views
Pharmacokinetic. Dr. Hayder B Sahib. *Half-life (plasma half-life) (t½): -The time required for the amount of drug to fall to 50% of an earlier measurement. -In 1st-order kinetic, (t½) is constant regardless of concentration.

E N D
Pharmacokinetic Dr. Hayder B Sahib
*Half-life (plasma half-life) (t½): • -The time required for the amount of drug to fall to 50% of an earlier measurement. • -In 1st-order kinetic, (t½) is constant regardless of concentration. • -(t½) is increased when the drug has high Vd, slow clearance and when there is high plasma –protein binding. • -(t½) is not indicative of duration of action and dosing schedule in the fallowing cases: • a-in drug which zero-order kinetic e.g. phenytoin • b-in drug with active metabolites e.g. diazepam. • c-hit and run drugs e.g. reserpine, organophospharous poisoning • d-presence of renal or hepatic dysfunction.
-(t½) is determined by volume of distribution(Vd) and clearance(CL) • 0.693× Vd • -(t½) = ------------------ (unit = time) • CL • -(t½) may be used to predict the manner in which plasma conc. alters in response to starting, altering or ceasing drug administ.
*Steady-state concentration: • - The condition in which the rate of drug elimination equals the rate of administration. • -When a drug is given at a constant rate(continuous or intermittent) the time to reach the steady-state depends only on the (t½) and, for all practical purposes, after 5 (t½) the amount of drug in the body will be constant and the plasma concentration will be at a plateau. • -Time to reach steady state for dobutamine((t½)=2 min) will be 10 min while for digoxin ((t½)= 36 hrs ) it will be more than 7.5 days.
*Volume of distribution(apparent) (Vd): • The ratio of the amount of drug in the body to the drug concentration in the plasma or blood. • Amount of drug in the body • Vd = ------------------------------------------ (unit=volume) • Plasma drug concentration • The calculated parameter for the Vd has no direct physical equivalent, therefore it is called apparent Vd. • -Drugs with low Vd (e.g. insulin=15, gentamicin=18) are mainly retained within plasma compartment, while drug with high Vd(digoxin=420, chloroquin=15000) are extensively bind to extravascular tissues.
И-Distribution; • Is the reversible movement of a drug between body compartment. • *Factors affecting drug distribution:- • 1-Ionization: unionized (lipid soluble) drugs diffused easier than ionized (water-soluble)drugs. • 2-Capillary permeability: • -In liver and spleen, the capillary are very leaky and drugs leave the capillaries regardless weather they are poorly-soluble, charged or polar. • -In other tissues, there is selective capillary permeability.
-Blood-brain barrier:- *Brain capillaries have tight junction. *Glucose and amino acids have specific carrier –mediated transport system. *Only lipophilic(lipid-soluble) drugs diffuse across brain capillaries (unless they are actively transported across) 3- Blood flow:- -Drugs reach the majority of tissues via general circulation . The rate at which drugs distribute from bloodstream into various tissues , depends on relative blood flow to the various tissues. -Brain, liver, kidneys, lungs > skeletal muscle> fat.
*Body-fluid compartments • -Total body water (TBW) = 42 L • -2 third of TBW in intracellular fluid space = 28L • -One third of TBW in extracellular fluid space =12L • -One third of extracellular fluid is intravascular i.e. plasma • -Adult blood volume ~ 5L
4-Plasma protein and tissue binding. • -Binding of a drug to plasma protein or a tissue compartment will tend to increase the drugs concentration in that compartment. • -E.g. warfarin is strongly bound to plasma albumin, which restrict warfarin diffusion out of the vascular compartment. • -Chloroquine is strongly bound to extravascular tissue proteins, which result in a marked reduction in plasma chloroquine. • -Examples of plasma protein binding of drugs • *warfarin 99% *diazepam 98% *tolbutamide 98% *phenytoin 91% *digoxin 25% *amoxicillin 18% *ethosuximide zero%
*Clearance (CL): • -The ratio of the rate of elimination of a drug to the concentration of the drug in plasma or blood. • Rate of elimination of drug • CL = ----------------------------------- unit=volume per unit time • Plasma drug concentration • -For a drug eliminated with 1st-order kinetic, clearance is constant, i.e. the ratio of the rate of elimination to plasma concentration is same regardless of plasma concentration.
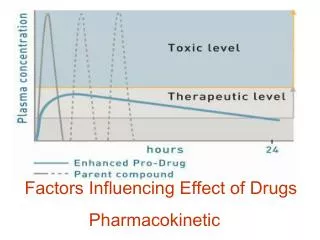
*Bioavailability: • -Is the fraction of administered dose that reaches the systemic circulation and it is 100% in case of I.V route. • - Bioavailability is reduced by; • -incomplete absorption • -1st-pass metabolism • -distribution to other tissues that occur before entering sys. Circulation. • -The area under the curve(AUC) is used to calculate the bioavailability • AUC route • Bioavailability= --------------------- • AUC IV
Individual pharmacokinetic process • 1-Absorption; • Absorption of drugs is depend on route of administration, blood flow and concentration of the drug at site of administration. • Routes of Drug Administration • Is determined primarily by:- • 1- The properties of the drug (e.g. water or lipid solubility, ionization) • 2-Theraputic objectives (e.g. the desirability of rapid onset or need for long-term administration or restriction to local site) • These routes include:- • A-Enteral e.g. oral • B-Parenteral e.g I.V • C-Others e.g inhalation
internal • 1-Oral :- Is the most common route of administration • *Advantages:- • - Convenience and acceptability. • *Disadvantages:- • - Absorption may be delayed , reduced or even enhanced after food • -Absorption may be slow or irregular after drugs that inhibit gut motility. • - Low bioavailability due to firs-pass metabolism • - Some drugs are not absorbed when given orally • - Some drug are destroyed in the gut
2-Subligual:- • *Advantages:- • - Quick effect is obtained. • - Bypass intestine and liver(avoid 1st pass metabolism) • *Disadvantages:- • - Inconvenience if use has to be frequent. • -Irritation of mucous membrane. • - Excessive salivation.
3-Rectal:- • *Advantages: • -Avoidance of stomach irritation. • - Suitable in vomiting. • - Suitable when cooperation is lacking (chidren). • - Partial avoidance of 1st pass metabolism. • *Disadvantages • -Psychological (embarrassment) • -Rectal inflammation (with repeated use) • -Absorption can be unreliable
B-Parenteral:- • 1-Intravascular:(most commonly I.V) • *Advantages: • - I.V route give highly predictable blood concentration • And allow rapid modification of dose. • -Suitable for drug that are not absorbed from gut or they Are irritant. • - With I.V route there is 100% bioavailability . • *Disadvantages: • -Dangerous if given quickly • -Local venous thrombosis as a result of prolong infusion. • -Introduce bacteria due to local contamination.
2-Intramuscular: • *Advantages: • -Reliable and suitable for irritant drug, depot preparations. • -Absorption is more rapid than subcutaneous injection. • -1st pass metabolism is avoided. • *Disadvantages: • -Not acceptable for self administration. • -It may be painful. • -Hematomas may occur when anticoagulant is given.
3-Subcuteanous: • *Advantages: • -Reliable and acceptable for self-administration. • - Avoid 1st pass metabolism. • *Disadvantages: • -Poor absorption in peripheral circulatory failure • -Lipo-atrophy due to repeated injection.
C-Others:- • 1-Inhalation:- • This route is particularly effective for patients with • respiratory complains( asthma or chronic obstructive air way diseases),because the drug is delivered directly to the site of action and systemic side effects are minimized. • 2-Topical: • -application to the skin • -application to the the eye, ear, nose,throat,airway or vagina. • The rate of absorption varies with area of application and the drugs formulations, but it usually slower than any of the routes listed previously. • .
3-Transdermal: • - Involves application to the skin for systemic effects • -Absorption usually occurs very slowly • -1st pass metabolism is avoided.
4-Intranasal: • Desmopressin is given intranasally in the treatment of diabetes insipidus • 5-Intrathecal: • Introduced drug directly in to CSF (e.g. amphotericin B in treating cryptococcal meningitis).
*Firs-pass effects (first-pass metabolism) • -Some drugs are ineffective when given orally e.g. nitroglycerin, insulin because of action of metabolizing enzymes in the intestinal wall and or liver, so very little or no drug reach general circulation and these drugs should not be given by oral route.
-Other drugs effected variably by 1st-pass effect. The oral dose of drugs that are effected extensively by 1st-pass effect (e.g. propranolol) should be much larger in comparison with parenteral route.