Download

1 / 33
330 likes | 484 Views
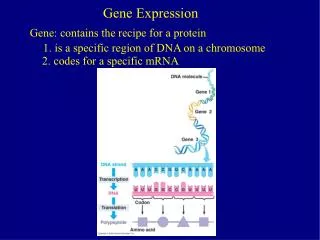
Estimating Gene Expression Signal For Affymetrix Arrays. Biostat 278 January 17 th , 2008. MAS 4.0. Expression Index: Average Difference Heuristic Present/Absent Call. Average Difference. Ave Diff is trimmed mean of PM i -MM i terms Removes >3 standard deviation values.

E N D
Estimating Gene Expression Signal For Affymetrix Arrays Biostat 278 January 17th, 2008
MAS 4.0 • Expression Index: Average Difference • Heuristic Present/Absent Call
Average Difference • Ave Diff is trimmed mean of PMi-MMi terms • Removes >3 standard deviation values
Problems with Ave-Diff • The removed values often removed important data • Negative values impossiblefor concentration or intensity
Background • Take into account the autofluorescence of array surface and NS binding • Compute average of 2% of lowest cell intensity values. • For P/A calls compute Q=(1/N) *∑(stdevi)/(√#pixelsi) *SF where the i corresponds to the background cell and the stddev is based on the pixels in the cell.
Present/Absent Call • Three types of calls: Absent, Marginal and Present based on three statistics. • Positive/Negative Ratio (3 / 4) • Positive: PM-MM>2*Q & PM/MM>1.5 • Negative: MM-PM>2*Q & PM/MM>1.5 • Positive Fraction (0.33 / 0.43) • Log Average Ratio (0.9 / 1.3)
MAS 5.0 • Expression Index: Tukey Biweight • Statistical Present/Absent Call
MAS 5.0 • A robust estimator, i.e. insensitive to outliers • Tukey proposed this function: • Signal is tukey biweight (log (PM-CT)) • CT is MM if MM<PM. • If MM>PM, CT = PM * tukey biweight estimate of log(PM/MM) ratio • If most MMs>PM for a probe, set CT = PM -
Rules for Computation Rule 1: If the Mismatch value is less than the Perfect Match value, then the Mismatch value is considered informative and the informative and the intensity value is used directly as an estimate of estimate of stray signal. Rule 2: If the Mismatch probe cells are generally informative across the probe set probe set except for a few Mismatches, an adjusted Mismatch value is used for uninformative Mismatches based on the on the biweight mean of the of the Perfect Match and Mismatch ratio. Rule 3: If the Mismatch probe cells are generally uninformative, the uninformative Mismatches are replaced with a value that that is slightly smaller than the Perfect Match. These probe sets are generally called Absent by the Detection algorithm
Tukey Biweight Estimator • X = signal • M = median(X) • S = median(|X-M|) = median of the absolute values of deviations from median • U = X-M/(5*S) = normalized distance from median • W = (1-U^2)^2 if U<1, W=0 else • T = Weighted mean of the signals:
At zero concentration PM has non-zero intensity As concentration increases, intensity increases
Average Signal for 12 human spiked transcripts (3x replicate)
Resistance to outliers • Introduce 10% artificial outliers to check robustness • Nonparametric correlation to handle both log-scale and linear-scale data • Verify data against known spike concentration
Detection Call • MAS 5.0 makes an absent/marginal/present call for each gene, called “detection” and provides a p value for that call. • Define R = (PM-MM)/(PM+MM) • R near 1 means PM>>MM • R near or below 0 means PM <= MM • Tau is cutoff for R (default 0.015) to be present • Use Wilcoxon signed rank test to determine if the Rs for an mRNA are higher than Tau. Report p value of this test.
Summary • A model for MAS 5.0 probe set intensity measure is • log( log(PMij - CTij) = log(θi) + εij ,where where j = 1,…,J. • The expression quantity on array i is represented with θi and εij is the error term which is equal to the variance for j =1,…,J.
MAS Normalization • Global Scaling: Multiplies the output of each chip by scaling factor to make average intensity equal to some arbitrary target intensity. • Intensity value is the average level of fluorescence over the entire chip. • All signals are scaled (identically) so the intensity value hits a target intensity • Target intensity must be fixed for chips to be comparable • Scaling factor is indicative: • High scaling factor = dim chip, indicating suspect data • Variable scaling factors within an experiment indicate variability in sample preparation and also suggest that the data is suspect. • What if different level of overall expression in the samples?
Li-Wong 2001(b) • Reduced model: Yij = PMij - MMij = θiφj + εij
Invariant Subset Normalization • Arrays have different overall brightness so they must be normalized. • There is a not necessarily a simple multiplicative relationship between samples • Ideally want to base normalization on the subset of genes that have constant expression between samples.
Invariant Subset Normalization • However, small group of control genes is insufficient to properly normalize entire range of expression. • Invariant Subset Normalization seeks to find the subset of probes that are do not change between samples.
Invariant Subset Normalization • Algorithm: 1. Compute ranking for all probes within each sample. 2. For each probe compute: PRD= (Rank1-Rank2)/ # probes 3. Retain probes with PRD <0.003 in low range and probes with PRD < 0.007 in high range. 4. Iterate procedure until the subset of retained probes does not change 5. Fit a piecewise linear normalization relationship between the set of invariant probes between the two samples.