Download

1 / 34
360 likes | 644 Views
The Role of Drug Metabolism Studies in Optimizing Drug Candidates. Kenneth Santone, PhD Bristol-Myers Squibb Metabolism and Pharmacokinetics / Pharmaceutical Candidate Optimization. ALTERNATE TITLE:. Why All the Chemist's Wonderful Compounds Don't Become Drugs!. Our Focus.

E N D
The Role of Drug Metabolism Studies in Optimizing Drug Candidates Kenneth Santone, PhD Bristol-Myers Squibb Metabolism and Pharmacokinetics / Pharmaceutical Candidate Optimization
ALTERNATE TITLE: • Why All the Chemist's Wonderful Compounds Don't Become Drugs!
Our Focus • Unmet medical need • First in class • Best in class • Need for efficiency and productivity enhancement
What are we faced with? • Industrialization of pharmaceutical research • Unprecedented increase in identification of targets • Corresponding increase in throughput of chemistry • Blurring of traditional discovery-development interface • Focus and emphasis on “developability”(early go/no go decisions) • Improve success rate • Reduce development timeline • Necessity for increasing efficiency and productivity
‘Old’ Model of Drug Discovery ‘New’ Model of Drug Discovery Validated Hits Hits Design Efficacy & Design Efficacy & PAT & Synthesis Selectivity Testing & Synthesis Selectivity Screening & Testing Predictions Lead Candidates Physicochemical, ADME Detailed Physicochemical, & Tox Workup ADME & Tox Workup Development Compound Development Compound Drug Discovery Paradigm Shift More informed decision making during Lead Optimization, through quicker and earlier evaluation of PAT attributes
The Hand-off from Drug Discovery to Development: The Top Ten Quotations We All Know and Love* 10. 9. 8. 7. 6. 5. 4. 3. 2. 1. “The molecular weight? 850. Why? Is that a problem?” “We’ll need eight different capsule strengths for Phase I.” “The compound is very potent in the in vitro screen but does not work well in the animal efficacy model.” “Now that you mention it, our solutions were a little cloudy.” “The compound is highly insoluble but Pharmaceutical Development will fix the problem.” “BMS-XXXXXX is a highly potent and selective inhibitor of (the target).In mouse models, the optimal dose was 200 mg/kg.” “Toxicity?! It’s not the drug; must be a metabolite unique to that animal species.” “Animal bioavailability ranged from 65% to <1%, depending on species.” “Gee, we didn’t have any problems when we gave it in DMSO.” “It’s a great compound, but it has formulation problems.” *why great compounds don’t always become drugs Partially adapted from R.A. Lipper
Critical Interfaces in Drug Discovery* Chemistry Biology Activity Safety Metabolism & Pharmacokinetics Pharmaceutics Optimized Compound *Analytical Chemistry (Bioanalysis) involved in every one of these disciplines
Role of ADME* Studies • Selection of quality drug candidate for development • Developability • First-in-class vs. best-in-class • Crisp go/no go decisions • Optimization of drug discovery and early development processes • Multi-tiered approach for ADME studies • Equal partnership with all functional areas Lead Discovery BiologyChemistry PharmaceuticsDrug Safety Analytical R&DClinical Pharmacology Process Chemistry • Blurring of traditional discovery-development interface * Absorption, Distribution, Metabolism, Excretion
Selection of Drug Candidates:Focus on Developability • Permeability • Transport • Metabolic stability • P-450 mediated drug interactions • PK/PD assessment • Distribution • Protein binding • Biopharmaceutics • Active/reactive/toxic metabolites • In vivoPK/bioavailabilityin animals • Prediction of PKand efficacious doses in humans
Tiered-Approach for ADME Studies • Hits to Lead • In vitro Studies • Permeability • P450 inhibition • Metabolic Stability • In silico predictions • Objective • Develop SAR • Chemotype selection
Tiered-Approach for ADME Studies • Lead Optimization • In vitro Studies • Permeability/transport • P450 inhibition • Metabolic Stability • Reaction phenotyping • Protein binding • In vivo PK • Cassette dosing • Individual PK • Tissue penetration • Early biotransformation • Objective • Identify a lead compound • Feedback to chemistry/biology
Tiered-Approach for ADME Studies • Lead Selection • Absolute bioavailability in pharmacology/toxicology models • Dose dependency in PK • Mechanism of absorption • Assess potential for DDI • Characterization of metabolites, routes of elimination • Assess formation of active metabolites • Interspecies differences in metabolism and in vitro-in vivo correlation • Extrapolation of ADME properties to man from in vitro and in vivo data • Determination of PK/PD relationships; help selection of doses for First in Human studies • Objective • Characterize the lead compound • Identify risks/opportunities
How In Vitro Metabolic Stability Relates to Clearance? TBC = CLhepatic + CLrenal + CLother CLhepatic = CLmetabolism + CLbiliary CLmetabolic = fB *CLintrinsic * Qh / fB *CLintrinsic + Qh well stirred model of organ extraction Intrinsic Clearance (CLi) = Vmax / Km = vo / Cu through rearrangement of the Michaelis-Menton eqn, assuming drug conc is < Km Depletion or Half-Life Method: CLi = (0.693 * liver wt) / (in vitro t1/2 * amount of liver)
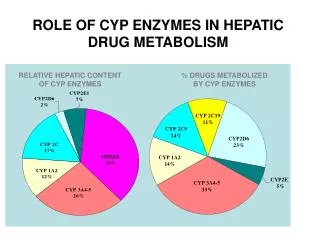
In Vitro Systems Liver microsomes high throughput and most common mostly oxidative (CYP & FMO) S9 fraction high throughput Phase I & Phase II metabolism Hepatocytes low throughput cell membrane/transporters intracellular concentration Phase I & Phase II metabolism In Vivo Animal Clearance In Silico Tools to Predict Metabolic Clearance In Vitro - In Vivo Correlation
Metabolic Stability to Select Compounds with Potentially Longer Half-Life Human Metabolic Stability: Microsome vs Hepatocyte 0.4 2 R = 0.8 0.3 Microsome Total Metabolic Rate 0.2 BMS:Y 0.1 0.0 -1 0 1 2 3 4 5 Hepatocyte Metabolic Rate • Lead compound is primarily glucuronidated in humans • Human in vitro systems with combination of oxidation and • glucuronidation employed for selection of back up
Major Reactions Involved in Drug Metabolism • OXIDATIVE REACTIONS (CYP, LM+NADPH) • N-Dealkylation: erythromycin, morphine, caffeine • O-Dealkylation: codeine, dextromethorphan • Aliphatic Hydroxylation: tolbutamide, midazolam • Aromatic Hydroxylation: phenytoin, amphetamine, warfarin • N-Oxidation: chlorpheniramine, dapsone • S-Oxidation: cimetidine, omeprazole • Deamination: amphetamine
Major Reactions Involved in Drug Metabolism • HYDROLYSIS REACTIONS (Esterase, ?LM+NADPH) • Ester Hydrolysis: aspirin, cocaine • Amide Hydrolysis: lidocaine, procainamide • CONJUGATION REACTIONS (Phase II, hepatocytes) • Glucuronidation: morphine, ibuprofen • Sulfation: acetaminophen • Acetylation: sulfonamides, isoniazid
Metabolic Stability Summary • Not all metabolism is hepatic. • Incubation concentration < Km balanced with assay sensitivity. • Need to correlate with in vivo model. • Fast in vitro clearance generally implies fast in vivo clearance, the reverse need not be true. • Confounding physical-chemical properties. • solubility, stability, purity, non-specific binding • Real concentration at enzyme active site? • protein binding, cell penetration, non-specific binding • In vitro systems generally underestimate CLi due to non-specific binding. • Can the stability be too good? Yes, in certain situations. • Many unknown factors to can contribute to a poor in vitro - in vivo correlation or poor estimation of human metabolic stability. • Nonetheless, in vitro methods are still the best method for predictions
Drug-Drug Interaction Summary • Major drug interactions are caused by either inhibition or induction of drug metabolizing enzymes. • Semi-quantitative predictions of drug interactions • many unknown factors • human ADME properties in vivo • Models provide numbers that must be placed in context with multiple factors: • therapeutic area • therapeutic index, route of administration • market competition • Animal models are not predictive of human interaction potential ??? • Static nature of in vitro systems compared to the dynamic in vivo system • Mixtures of interaction mechanisms from the same compound are extremely difficult to predict: • reversible + irreversible inhibition • inhibition + induction
Assessment of Active Metabolites • Issue • Similar metabolism and in vitro activity profile but different in vivo • activity profile • Apparent PK/PD disconnect • Solution • Rapid in vitro metabolism and biological activity assays
Assessment of Active Metabolites • Structural identification of active metabolites • MS/MS indicated presence of monohydroxylation • NMR showed site of hydroxylation • Subsequent steps • Monohydroxylated metabolite synthesized • Activity and PK properties confirmed
Assessment of Reactive Metabolites • A number of functional (chemical) elements have been associated with problems in drug discovery leading to toxicity • Metabolic activation to reactive intermediates • Interference with metabolic processes • Clinical manifestations include (preclinical measure) • Cellular (hepatic) necrosis (animal toxicity) • Idiosyncratic toxicity (glutathione adducts, protein covalent binding, immunogenic response) • Drug-drug interactions (mechanism-dependent CYP inhibition)
Examples of Reactive Metabolites • Furans • Furan substructure is associated with toxicity (eg. aflatoxin) and with CYP inhibition (eg. bergamottin)
Examples of Reactive Metabolites • Thiophenes • Thiophene substructure has been associated with several types of toxicity (predominately hepatotoxicity). Other thiophene containing drugs: ticlopidine, clopidigrel, raloxifene.
Examples of Reactive Metabolites • Anilines, Nitroaromatics • Anilines are associated with a number of types of toxicity (eg. methemoglobinemia, skin rashes, etc.). Nitroaromatics are primarily activated by initial reduction, often in the gut, followed by N-oxidation. • Anilines of polycyclic aromatic systems are often potent mutagens and carcinogens (eg., naphthylamine, aminofluorene) through conjugation of the hydroxylamine and subsequent loss of the conjugate to leave a nitrenium ion.
Examples of Reactive Metabolites • Amines, alkylamines • The metabolism of amines or alkylamines is generally related to time-dependent inhibition of CYP enzymes, with the nitroso species forming a tight complex with the heme iron, known as a MI complex. Other compounds that undergo this type of transformation and inhibit CYPs are TAO, erythromycin and verapamil
Examples of Reactive Metabolites • Quinone, Quinoid • Quinone-like compounds can exert their effects through direct alkylation of nucleophiles or through redox cycling between their oxidized and reduced forms
Examples of Reactive Metabolites • Acetylenes • Acetylenes have been found to be time-dependent inhibitors of CYP enzymes.
Examples of Reactive Metabolites • Acyl glucuronidation formation • Acyl glucuronides have been implicated in both direct hepatic damage and idiosyncratic toxicities
Challenges and Opportunities • HTS screens for prediction of permeability, metabolic stability, metabolic reactivity and DDI • How are we using these data? • Retrospective analysis on return of investment • The numbers in gray zone! • Secondary assays for better predictability • Application of animal PK/bioavailability data for lead optimization • Adequacy of permeability and metabolic stability data • Animals vs. humans: quantitative and qualitative differences in ADME properties • Informed decision based on drug metabolism and pharmacokinetic data • Low bioavailability vs. oral efficacy • Role of metabolite(s), reactivity of metabolite(s) • Protein binding • In vitro- in vivo correlation in animals and extrapolation to humans • Issue of enzyme induction in humans • In-vitro models and predictability • False and real alarm from in-vivo animal data
Challenges and Opportunities • Use of biomarkers • In-vivo biology, animals vs. humans • Development and validation of assays • Transfer from preclinical to clinical laboratories • Biomarkers = Surrogate marker = Efficacy/Toxicity • A balancing act of emerging science • The feedback loops • To and from chemistry • To and from biology • To and from drug safety • To and from pharmaceutics • To and from clinical pharmacology • Volume of data • Conversion of information into knowledge • Timing and availability
A Focused Application of ADME Studies • Active involvement earlier in the Discovery Process • Timely guidance to Chemistry to select chemotypes with desirable ADME properties • Maximize informed decision making during Lead Optimization • Improved ability to predict human metabolism and pharmacokinetics • Stronger partnerships with Drug Discovery and all areas of Pharmaceutical Development
Our Mission To ensure that no development candidate fails in the clinic due to an unforeseen metabolic or pharmacokinetic property
Acknowledgements David Rodrigues and Griff Humphreys • And finally …. Saeho Chong, Punit Marathe, Wen Chyi Shyu and Mike Sinz