Download

1 / 61
620 likes | 664 Views
Explore the evolution from traditional cDNA library sequencing to modern RNA-Seq technologies, providing a historic perspective, strategies for read mapping vs. de novo assembly, and insights into genomics and transcriptomics. Discover the power of de Bruijn Graph and Inchworm Algorithm in transcriptome reconstruction.

E N D
RNA Sequencing and transcriptome reconstruction Manfred G. Grabherr
A historic perspective • Traditional: sequence cDNA libraries by Sanger • Tens of thousands of pairs at most (20K genes in mammal) • Redundancy due to highly expressed genes • Not only coding genes are transcribed • Poor full-lengthness (read length about 800bp) • Indels are the dominant error mode in Sanger (frameshifts)
A historic perspective • Quantification: microarrays • Sequences have to be known • Annotations are often incomplete • No novel transcripts • Hybridization bias (SNPs) • Noise
Next-Gen Sequencing technologies • 1 Lane of HiSeq yields 30GB in sequence • Short reads (100nt), but: • Good depth, high dynamic range • Full-length transcripts • Novel transcripts • Allow for expression quantification • Error patterns are mostly substitutions • Strand-specific libraries
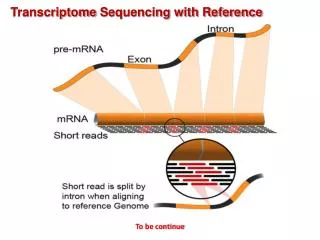
Strategy: read mapping vs. de novo assembly Haas and Zody, Nature Biotechnology 28, 421–423 (2010)
Strategy: read mapping vs. de novo assembly Good reference No genome Haas and Zody, Nature Biotechnology 28, 421–423 (2010)
Leveraging RNA-Seq for Genome-free Transcriptome Studies Brian Haas
A Paradigm for GenomicResearch WGS Sequencing Assemble Draft Genome Scaffolds Methylation Tx-factorbinding sites SNPs Proteins
A Paradigm for GenomicResearch RNA-Seq WGS Sequencing Assemble Align Draft Genome Scaffolds Transcripts Methylation Tx-factorbinding sites Expression SNPs Proteins
A Maturing Paradigm for TranscriptomeResearch RNA-Seq WGS Sequencing Assemble Align Assemble Draft Genome Scaffolds Transcripts Methylation Tx-factorbinding sites Expression SNPs Proteins
A Maturing Paradigm for TranscriptomeResearch RNA-Seq $$$$$ $$$$$ $$$$$ $$$$$ WGS Sequencing + $ Assemble Align Assemble Draft Genome Scaffolds $ Transcripts Methylation Tx-factorbinding sites Expression SNPs Proteins
A Maturing Paradigm for TranscriptomeResearch RNA-Seq $$$$$ $$$$$ $$$$$ $$$$$ WGS Sequencing + $ Assemble Align Assemble Draft Genome Scaffolds $ Transcripts Methylation Tx-factorbinding sites Expression SNPs Proteins
A Maturing Paradigm for TranscriptomeResearch RNA-Seq $$$$$ $$$$$ $$$$$ $$$$$ WGS Sequencing + $ Assemble Align Assemble Draft Genome Scaffolds $ Transcripts Methylation Tx-factorbinding sites Expression SNPs Proteins
A Maturing Paradigm for TranscriptomeResearch RNA-Seq $$$$$ $$$$$ $$$$$ $$$$$ WGS Sequencing + $ Assemble Align Assemble Draft Genome Scaffolds $ Transcripts Methylation Tx-factorbinding sites Expression SNPs Proteins
De-novo transcriptome assembly Brian Haas Moran Yassour Kerstin Lindblad-Toh Aviv Regev NirFriedman David Eccles AlexiePapanicolaou Michael Ott …
The problem Transcript
The problem Transcript Reads
The problem Transcript Reads Assembly Transcript
The problem Transcript Paralog A Paralog B Reads Assembly Transcript
The problem Transcript Isoform A Isoform B Reads Assembly Transcript
Transcriptome vs. Genome assembly • Genome: • Large • High coverage • Long mate pairs (hard to make) • Linear sequences • Even coverage • Transcriptome: • Smaller • Standard paired-end Illumina (1 lane) • Multiple solutions (alternative splicing) • Uneven coverage (expression)
Transcriptome vs. Genome assembly • Genome: • Large • High coverage • Long mate pairs (hard to make) • Linear sequences • Even coverage • Transcriptome: • Smaller • Standard paired-end Illumina (1 lane) • Multiple solutions (alternative splicing) • Uneven coverage (expression) • In common: k-mer based approach
The k-mer • K consecutive nucleotides Reads K-mers Graph
The de Bruijn Graph • Graph of overlapping sequences • Intended for cryptology • Fixed length element: k • CTTGGAA • TTGGAAC • TGGAACA • GGAACAA • GAACAAT
The de Bruijn Graph • Graph has “nodes” and “edges” • G GGCAATTGACTTTT… • CTTGGAACAAT TGAATT • A GAAGGGAGTTCCACT…
Iyer MK, Chinnaiyan AM (2011) Nature Biotechnology29, 599–600
Iyer MK, Chinnaiyan AM (2011) Nature Biotechnology29, 599–600
Iyer MK, Chinnaiyan AM (2011) Nature Biotechnology29, 599–600
Iyer MK, Chinnaiyan AM (2011) Nature Biotechnology29, 599–600
Inchworm Algorithm Decompose all reads into overlapping Kmers (25-mers) Identify seed kmer as most abundant Kmer, ignoring low-complexity kmers. Extend kmer at 3’ end, guided by coverage. G A GATTACA 9 T C
Inchworm Algorithm G 4 A GATTACA 9 T C
Inchworm Algorithm G 4 A 1 GATTACA 9 T C
Inchworm Algorithm G 4 A 1 GATTACA 9 T 0 C
Inchworm Algorithm G 4 A 1 GATTACA 9 T 0 C 4
Inchworm Algorithm G 4 A 1 GATTACA 9 T 0 C 4
Inchworm Algorithm G A 0 5 T 1 G C 4 0 A 1 GATTACA 9 T 0 G C 1 4 A 1 T C 1 1
Inchworm Algorithm G A 0 5 T 1 G C 4 0 A 1 GATTACA 9 T 0 G C 1 4 A 1 T C 1 1
Inchworm Algorithm A 5 G 4 GATTACA 9
Inchworm Algorithm A 5 C G 0 4 T 0 GATTACA A 9 6 G 1
Inchworm Algorithm A 5 G 4 GATTACA A 9 6 A 7 Report contig: ….AAGATTACAGA…. Remove assembled kmers from catalog, then repeat the entire process.
Inchworm Contigs from Alt-Spliced Transcripts=> Minimal lossless representation of data +
Chrysalis Integrate isoforms via k-1 overlaps
Chrysalis Integrate isoforms via k-1 overlaps
Chrysalis Integrate isoforms via k-1 overlaps Verify via “welds”
Chrysalis Integrate isoforms via k-1 overlaps Verify via “welds” Build de Bruijn Graphs (ideally, one per gene) Build de Bruijn Graphs (ideally, one per gene)