Download

1 / 42
420 likes | 545 Views
Probabilistic Techniques for the Clustering of Gene Expression Data. Speaker: Yujing Zeng Advisor: Javier Garcia-Frias Department of Electrical and Computer Engineering University of Delaware. Contents. Introduction Problem of interest Introduction on clustering

E N D
Probabilistic Techniques for the Clustering of Gene Expression Data Speaker: Yujing Zeng Advisor: Javier Garcia-Frias Department of Electrical and Computer Engineering University of Delaware
Contents • Introduction • Problem of interest • Introduction on clustering • Integrating application-specific knowledge in clustering • Gene expression time-series data • Profile-HMM clustering • Integrating different clustering results • Meta-clustering • Conclusion
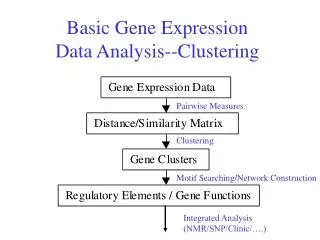
Gene Expression Data DNA (Gene) measure Messenger RNA (mRNA) Transcription Regulation Protein Translation • The pattern behind these measurements reflects the function and behavior of proteins
Clustering Gene Expression Data (cont.)
Clustering Gene Expression Data (cont.)
Clustering What Is Clustering? Clustering can be loosely defined as the process of organizing objects into groups whose members are similar in some way. • All clustering algorithms assume the pre-existence of groupings among the objects to be clustered • Random noise and other uncertainties have obscured these groupings
Clustering Advantages of Clustering • Unsupervised learning • No pre-knowledge required • Suitable for applications with large database • Well-developed techniques • Many approaches developed • Vast literature available
Clustering Problem of Interest • Difficult to integrate information resources other than the data itself • Pre-knowledge from particular applications • Clustering results from other clustering analysis
Clustering Profile-HMM clustering - exploiting the temporal dependencies existing in gene expression time-series data
Clustering Gene Expression Time-Series Data • Special property • Horizontal dependencies: dependence existing between observations taken at subsequent time-points • Similarity between a pair of series is decided by their patterns across the time axis • Gene expression time-series data • Collected by a series of microarray experiments implemented in consecutive time-points • Each time sequence representing the behavior of one particular gene along the time axis
Clustering Hidden Markov Models to Model Temporal Dependencies • Hidden Markov models (HMMs) are one of the most popular ways to model temporal dependencies in stochastic processes (speech recognition) • Characterized by the following parameters: • Set of possible (hidden) states • Transition probabilities among states • Emission probability in each state • Initial state probabilities • Doubly stochastic structure allows flexibility in the modeling of temporal dependencies S1 S2
Previous Work Clustering • Limited quality of the resulting HMM because of small training set (one series for each HMM) • Lack of models for the whole data structure • Generate one HMM per gene • HMM-based distance [Smyth 97] • HMM-based features [Panuccio et al 02] • Generate one HMM per cluster • Autoregressive models (CAGED) [Ramoni et al 02] • HMM based EM clustering [Schliep et al 03] • Separate training for the model of each cluster • Requirement of additional technique to predict the number of clusters • Stationary assumption on the temporal dependencies
. . . S1T S12 S11 . . . . . . . . . . . . . . . SmTT Sm22 Sm11 Profile-HMM Clustering Clustering • Left-to-right model with each group of states associated with a time point • Only transitions among consecutive layers are allowed • Time dependencies at different times modeled separately • For each state, emission defined by a Gaussian density Each path describes a pattern in a probabilistic way.
Clustering Profile-HMM Clustering (cont.) • Similarity between two time series defined according to the probability that they are related to the same stochastic pattern • Training: To find the most likely set of patterns characterizing all the observed time series • Clustering: Group together the time series (genes) that are most likely to be related with the same pattern ( which corresponds a cluster Baum-Welch Viterbi
Profile-HMM Clustering (cont.) Clustering • Single HMM models the overall distribution of the data, so that the representative patterns (clusters) are selected simultaneously • As opposed to other HMMs approaches each stochastic pattern is built according to both positive and negative samples • Number of clusters is obtained automatically • Proposed model can be seen as a high dimensional self- organized network • Number of clusters is relatively stable with respect to number of states • Training and clustering procedures are standard techniques Easy implementation
Clustering Experiment Results: Dataset • Study on the transcriptional program of sporulation in budding yeast [Chu et al, 98] • Measures at 7 uneven intervals • Subset of 477 genes with over-expression behavior during sporulation • Original paper distinguishes 7 temporal patterns by visual inspection and prior studies)
Experiment Results:Number of Clusters from Proposed HMM Clustering • Same number of states at each time point, m • # of clusters is automatically determined by the HMM • Resulting # of clusters (and clustering structure) is relatively stable with respect to the number of states in the model • m=3 37=2187 possible patterns, but 12 resulting clusters • m=50350=7.8x1011 possible patterns, but 19 resulting clusters
Clustering Clustering Validation
Experiment Results: Comparison with Original Model Clustering • HMM increases the number of clusters from the original 7 to 16 • HMM identifies patterns mixed in the same original group and assign them into different clusters • Original metabolism group shows some inconsistent profiles • HMM refines this subset into 2 more consistent clusters
Experiment Results:Comparison with Other Clustering Methods Clustering • Compare with K-means and single-linkage with #clusters=16 • 14 out of 16 clusters in single-linkage are singletons Despite DB and separation indices, real patterns are not described in the single-linkage clusters
Summary for HMM Clustering Clustering • A novel HMM clustering approach proposed to exploit the temporal dependencies in microarray dynamic data • HMM performance evaluated using data studying the transcriptional program of sporulation in budding yeast • HMM capable of identifying a reasonable number of clusters, stable with model complexity, without any a priori information • Evaluation indices show that HMM provides a better description of the data distribution than other clustering techniques • Biological interpretation from the HMM results provides meaningful insights
Clustering Problem of Interest • Difficult to integrate information resources other than the data itself • Pre-knowledge from particular applications • Clustering results from other clustering analysis
Clustering Meta-clustering - integrating different clustering results
Clustering Facing Various Clustering Approaches… • There is no single best approach for obtaining a partition because no precise and workable definition of ‘cluster’ exists • Clusters can be of any arbitrary shapes and sizes in a multidimensional pattern space • Each clustering approach imposes a certain assumption on the structure of the data • If the data happens to conform to that structure, the true clusters are recovered
Result of K-means Result of SOM Result of Single-linkage Result of SOM Result of K-means Clustering Example of Clustering
Result of K-means Result of SOM Result of K-means Result of Single-linkage Result of Single-linkage Clustering Example of Clustering(cont.)
Clustering Problem of Interest • Difficult to evaluate, compare and combine different clustering results • Different cluster sizes,boundaries, … • High dimensionality • Large amount of data • Although many clustering tools are available, there are few to extract the information by comparing or combining two or more clustering results
Clustering Proposed Approach • An adaptive meta-clustering approach • Extracting the information from results of different clustering techniques • And combining them into a single clustering structure • - so that a better interpretation of the data distribution can be obtained
Alignment Combination Meta-clustering Clustering Adaptive Meta-clustering Algorithm
Clustering Dc Matrix • nn matrix, where n is the size of the input data set • Each entry Dc(i,j) is the cluster-based distance between data point i and j • The cluster-based distance, which we define, shows the dissimilarity between every two points
Cluster I P vectors Cluster-Based Distance X5 Cluster IV X3 X4 X6 X7 X1 X2 Cluster II Cluster III Clustering Cluster-Based Distance
Clustering Combination • Assume that is a clustering structure that we want to discover from the input dataset. Let denote the corresponding matrix of cluster-based distances (Dc) • Given a pool of clustering results, we can estimate as
Merging criteria Clustering Meta-Clustering • Using agglomerative hierarchical approach
Clustering Simulation Results
Clustering Simulation Results (cont.)
Clustering Simulation Results (cont.) Single-linkage K-means SOM Meta-clustering
Clustering Simulation Results (cont.) • Yeast cell-cycle data Karen M. Bloch and Gonzalo Arce, “Nonlinear correlation for the Analysis of Gene Expression Data”, ISMB 2002.
Size Percentage of profiles in the group that are from the function class Percentage of profiles in the function class that are contained in the group Chromatin Structure Glycolysis Protein Degradation Spindle Pole Chromatin Structure Glycolysis Protein Degradation Spindle Pole Average-linkage 1 8 100% 100% 2 1 100% 6% 3 16 100% 94% 4 40 73% 27% 100% 100% SOM 1 8 100% 100% 2 15 100% 88% 3 31 3% 94% 3% 6% 100% 9% 4 11 9% 91% 6% 91% K-means 1 11 73% 27% 100% 10% 2 13 100% 76% 3 26 100% 90% 4 15 27% 73% 24% 100% Meta-Clustering 1 8 100% 100% 2 17 100% 100% 3 30 97% 3% 100% 9% 4 10 100% 91% Clustering Simulation Results (cont.)
Chromatin structure Glycolysis Protein degradation Spindle Pole
Clustering Summary for Meta-Clustering • The evaluation and combination of different clustering results is an important open problem • The problem is addressed by • Defining a special distance measure, called Dc, to represent the statistical "signal" of each cluster • Combining the information together in a statistical way to form a new clustering structure • The simulations show the robustness of the proposed algorithm
Conclusion • We are interested on analyzing gene expression data sets and inferring biological interactions from them • The study is focused on clustering • Including the pre-knowledge in clustering process • Integrating different clustering results • The future work will give more emphasis on real applications