Download

1 / 13
170 likes | 419 Views
Molecular diagnostics in congenital adrenal hyperplasia (21-hydroxylase deficiency ). Introduction.

E N D
Molecular diagnostics in congenital adrenal hyperplasia (21-hydroxylase deficiency)
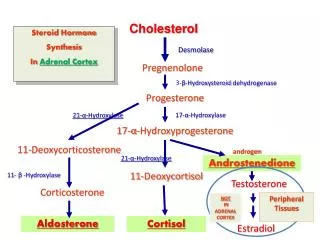
Introduction • Congenital adrenal hyperplasia (CAH), the inherited inability to synthesize cortisol, is one of the most common inherited endocrine disorders. Cortisol is normally synthesized in the zonafasciculata of the adrenal cortex in five enzymatic steps: cleavage of the cholesterol side chain to convert cholesterol to pregnenolone, dehydrogenation at the 3 β position to yield progesterone and a series of hydroxylations at the 17α, 21 and 11 βpositions to form 17α-hydroxyprogesterone, 11-deoxycortisol and cortisol respectively. Typical signs of androgen excess include masculinization of female external genitalia and precocious pseudopuberty in both sexes. Patients undergo rapid somatic growth with premature epiphyseal closure resulting in short adult stature. Moreover, elevated metabolites with mineralocorticoid activity, such as deoxycorticosterone and its derivatives, cause hypertension in about two thirds of patients. Patients are treated with glucocorticoid replacement and with antihypertensive therapy if necessary.
Congenital adrenal hyperplasia (CAH) is inherited as an autosomal recessive disorder and maps within the MHC locus on chromosome 6. Indeed, linkage analysis using HLA Class I and or Class II markers is often used for genetic studies of suitable families. About 95% of the cases of CAH are due to a limited number of mutations in the 21-hydroxylase gene (21-OH or CYP21 - also known as P450C21B). Molecular analysis of CAH is complicated due to the presence of a non-functional pseudogene, CYP21P - also known as P450C21A) adjacent to the functional 21-OH gene. Most of the pathogenic CYP21 mutations are found in the pseudogene, hence, it is assumed that gene conversion is a common mutational mechanism in CAH (see below). The other significant cause of CAH is gross deletion of the functional 21-OH gene. Presumably the presence of the tandem homologous CYP21 and CYP21P genes provides the opportunity for unequal crossing over events during meiosis.
CompGene's strategy for molecular diagnosis of CAH first uses a Southern blot analysis with a 21-OH cDNA probe and the restriction endonucleases Taq I and a double digest with Eco RI/Bgl II. As shown in the cartoon above, distinct fragments and fragment dosage patterns will be revealed in the presence of deletions and gene conversions. An example of Southern blot analysis of the 21-hydroxylase locus using the restriction enzyme Taq I is shown below.
A normal individual's DNA hybridization pattern is shown in lane 1. All other lanes show individuals affected by CAH and illustrates the limited information that is obtained by this diagnostic approach alone. Although the patient in lane 2 obviously has homozygous deletion of the 21-hydroxylase locus as the cause of his disease, the other patients show subtle or no obvious changes. For example, the patients in lanes 5, 7 and 8 appear completely normal, the patient in lane 4 shows a half dose of the 3.7 KbpTaq I fragment consistent with gene deletion/conversion, while the patient in lane 6 shows an increased copy number of the pseudogene as detected by increased intensity of the 3.2 KbpTaq I fragment.
Our method of evaluation of 21-hydroxylase mutations uses either "amplification refractory mutation analysis" where a PCR product is obtained when a specific nucleotide is present (either normal or mutant) in the 21-hydroxylase gene (P30L, A/C656G, exon 3 deletion, I172N, exon 6 cluster, R356W) or, in some cases, uses a specific restriction endonuclease to detect the sequence change that results in the loss of a restriction endonuclease site (Q318X, V281L). Examples of direct mutation analysis are shown in the figures below. n this first figure, the upper panel shows PCR results with no DNA controls in lanes 1 and 6, and test samples in the rest of the lanes. Lanes 1- 5 test for the wild type 172 T nucleotide while lanes 6 - 10 test for the mutant 172 A nucleotide that results in the I172N mutation. As can be seen by the results depicted in lanes 5 and 10, this individual is a carrier of the I172 mutation. All other individuals are wild type at this site (although they might be deletion carriers on the other chromosome).
The lower panel illustrates the use of a restriction endonuclease to detect a point mutation. Specific restriction fragments appear in the presence of the mutation and are indicated by the red arrows. Lane 4 shows an individual who carries the V281L mutation on one chromosome while the individuals in lanes 7 and 9 carry the Q318X mutation
Similar analysis shows in the upper portion of the figure shown below, detection of the R356W mutation in the samples in lanes 8, 9 and 10. The lower portion of the figure shows detection of the common A/C656G splice mutation in the samples of lanes 7, 8 and 9.
In certain families, it may be necessary to consider a linkage analysis study. An example of this type of analysis is shown below.
In this fully informative situation, the proband has inherited the paternal C allele and maternal A allele at D6S276 and the paternal C allele and maternal A allele at D6S1568. DNA prepared from the CVS obtained from the mother showed a recurrence of the predicted affected haplotypes. In addition, the evaluation of these flanking markers confirmed fetal cell sampling by the CVS with no evidence of maternal cell contamination. Wherever possible, CompGene performs evaluation of flanking markers to complement direct mutation analysis studies. These and our other in-house flanking and physically mapped STR markers provide >99% accuracy in linkage analysis situations. Please consult with Celtek when linkage studies are being considered. We are often asked about genotype/phenotype correlations. This is a difficult question to answer because CAH can present in a variety of clinical forms presumably due to the nature of the CYP21 mutations present and compound heterozygosity
These clinical presentations range from the life-threatening "classic" or salt wasting presentations in infancy to late-onset "non-classic" forms that may present simply as mild hirsutism in females (for examples of mutation analysis in the latter group, see Blanche et al. (1997) Hum. Genet. 101:56-60. It is also possible that other genotypic factors not at the 21-hydroxylase locus can modify clinical severity. In a general sense, the figure below modified from Wedell et al. (1996; Clin. Lab. Med. 16:125-137) illustrates the spectrum of phenotypes associated with particular 21-hydroxylase mutations.