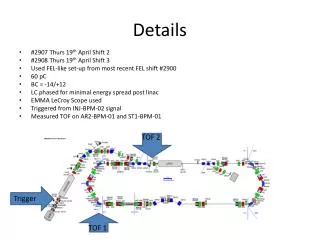
Download

1 / 33
330 likes | 343 Views
This study aims to assess the efficacy of trametinib in patients with recurrent or progressive low-grade serous ovarian cancer or peritoneal cancer. The study is conducted in accordance with international guidelines and protocols.

E N D
LOGS: A randomised phase II/III study to assess the efficacy of trametinib(GSK 1120212) in patients with recurrent or progressive low grade serous ovarian cancer or peritoneal cancer (GOG-0281) (EudraCT Number: 2013-001627-39) UK Chief Investigator – Professor Charlie Gourley UK Sponsor – NHS Greater Glasgow & Clyde and University of Glasgow UK Co-ordinating Centre: CRUK Clinical Trials Unit, Glasgow Pharmacy Initiation Slides – Version 1.0 25th March 2015 LOGS Pharmacy Initiation Slides v1.0 25_03_15
Study Details • Study will be conducted according to ICH GCP guidelines • Study conducted in accordance with the EU Directive 2001/20/EC • Trial carried out in accordance with the World Medical Association Declaration of Helsinki (1964) and the Tokyo (1975), Venice (1983), Hong Kong (1989), South Africa (1996), Edinburgh (2000), Washington (2002), Tokyo (2004), Seoul (2008) amendments **Please note this presentation has been prepared as part of your site initiation. These slides are a compliment to the protocol and UK appendix to protocol, all site staff must have read and understood the protocol, UK appendix and the study requirements prior to signing off the initiation acknowledgement sheet.** LOGS Pharmacy Initiation Slides v1.0 25_03_15
Study Team in UK UK Chief Investigator: Professor Charlie Gourley Lead Pathologist UK: Dr David Millan Project Manager: Karen Carty Pharmacovigilance: Lindsey Connery Sponsor Pharmacy Contacts: Paula Morrison + Eliza Valentine Clinical Trial Co-ordinator: Diann Taggart Clinical Trial Monitor: Jan Graham 3 LOGS Pharmacy Initiation Slides v1.0 25_03_15
Pharmacy Initiation • Protocol and Treatment overview • IMP Presentation and Management • LOGS site file and documentation • Site initiation process LOGS Pharmacy Initiation Slides v1.0 25_03_15
LOGS Protocol and treatment overview LOGS Pharmacy Initiation Slides v1.0 25_03_15
Study Design Design: This is an un-blinded, randomized phase II/III study comparing trametinib to “standard therapy” (consisting of one of five commercially available agents) in patients with low-grade serous carcinoma of the ovary or peritoneum previously treated with platinum based chemotherapy in women with recurrent low grade serous. Primary Objective: To estimate the progression-free survival (PFS) hazard ratio of trametinib compared to that of “commercially available therapies” consisting of one of five commercially available agents in women with recurrent low grade serous carcinoma of the ovary or peritoneum previously treated with platinum-based chemotherapy. 6 LOGS Pharmacy Initiation Slides v1.0 25_03_15
Treatment and Duration In each arm of the study, one cycle is 28 days. The first dose of study medication should be administered as close as possible after randomization. Arm A (Control Arm) Clinician’s choice of ‘control’ arm is made from the list below prior to randomization: Letrozole2.5mg orally once daily continuous treatment until progression or unacceptable toxicity Tamoxifen 20mg orally twice daily continuous treatment until progression or unacceptable toxicity Paclitaxel 80mg/m2 IV infusion over one hour on days 1, 8, and 15 of a 28 day cycle until progression or unacceptable toxicity or until 6 cycles have been administered* Pegylated Liposomal Doxorubicin 40 or 50mg/m2 IV infusion over one hour on day 1 every 28 days until progression or unacceptable toxicity or until 6 cycles have been administered* Topotecan 4.0 mg/m2 IV infusion over 30 minutes on days 1, 8, and 15 of a 28 day cycle until progression or unacceptable toxicity or until 6 cycles have been administered* *more than 6 cycles of chemotherapy can be administered at investigator’s discretion Arm B (Experimental Arm) Trametinib2 mg orally once daily continuous treatment until progression or unacceptable toxicity 7 LOGS Pharmacy Initiation Slides v1.0 25_03_15
Crossover If a patient develops progressive disease in Arm A (Control Arm - as defined in study protocol Section 8.134), the patient will be given the opportunity to crossover to Arm B (Experimental Arm). • Prior to crossover, the following must occur: • The patient’s progression must be fully documented on the relevant GOG electronic case report forms (CRFs) and submitted via Medidata Rave Electronic Data Entry System (www.imedidata.com) online application which is being used for the study. The relevant CRFs require to be submitted prior to the patient starting crossover treatment. • All eligibility criteria as defined in study protocol section 3 must be met (with the exception of 3.143, 3.15, and 3.114). This includes requirement that 4 weeks must elapse between the end of treatment with Arm A and start of treatment on Arm B. These requirements will be documented on the CRFs. • If the patient meets all of the above noted requirements, she will be able to crossover and commence treatment on Arm B. LOGS Pharmacy Initiation Slides v1.0 25_03_15
Key Inclusion Criteria (1) Patients will be eligible for the study if the following criteria are met: • Patients aged 18 years of age or older with the following tumours: • Patients initially diagnosed with low-grade serous ovarian or peritoneal carcinoma that recur as low-grade serous carcinoma (invasive micropapillary serous carcinoma or invasive grade I serous carcinomas as defined by GOG, FIGO WHO or Silverberg). • Patients initially diagnosed with serous borderline ovarian or peritoneal carcinoma that recur as low-grade serous carcinoma (invasive micropapillary serous carcinoma or invasive grade I serous carcinomas as defined by GOG, FIGO WHO or Silverberg). • At least 4 weeks must have elapsed since the patient underwent any major Surgery • Patients must have documented low-grade serous carcinoma. • All patients must have measurable disease as defined by RECIST 1.1. • Prior therapy : - Patients must have recurred or progressed following at least one platinum-based chemotherapy regimen. - Patients may have received an unlimited number of prior therapy regimens. - Patients may not have received all of the five choices in the “standard therapy” arm. - Any hormonal therapy directed at the malignant tumor must be discontinued at least one week prior to registration - Any other prior therapy directed at the malignant tumor, including chemotherapy and radiation therapy, must be discontinued at least 4 weeks prior to registration. Any investigational agent must be discontinued at least 28 days prior to registration. See study protocol for full inclusion/exclusion criteria LOGS Pharmacy Initiation Slides v1.0 25_03_15
KeyInclusion Criteria (2) • Women of child-bearing potential and men must agree to use a highly effective method of contraception prior to study entry, during the study participation, and for six months after the last dose of the drug. Women of child-bearing potential must have a negative serum pregnancy test within 14 days prior to randomization, cannot be breast-feeding, and must agree to use a highly effective form of contraception throughout the treatment period and for 6 months after the last dose of study treatment. • Patients must have signed an approved informed consent. • Patient must have a performance status of 0 or 1. • Patients must have ability to swallow and retain orally administered medication.. • All prior treatment-related toxicities must be CTCAE v4 grade <1 (except alopecia) at the time of randomization. See study protocol for full inclusion/exclusion criteria LOGS Pharmacy Initiation Slides v1.0 25_03_15
Key Inclusion Criteria (3) • Patients must have a left ventricular ejection fraction > lower limit of normal by ECHO or MUGA • Patients must have adequate: - Bone Marrow Function: Absolute neutrophil count (ANC) > to 1.5 x 109/l Platelets > 100 x 109/l, Haemoglobin > 9.0g/dl - Renal Function: Serum creatinine < 1.5 mg/dl OR calculated creatinine clearance (Cockroft-Gault formula) > 50ml/min or 24 hour urine creatine clearance > 50ml/min - Hepatic Function: Bilirubin < 1.5 X ULN, ALT < 2.5 X ULN, AST < 2.5 X ULN, Albumin > 2.5 - Coagulation: PT and APTT <1.5 X ULN **All samples must be taken within 7 days prior to treatment** • If letrozole is selected as the control therapy, patients must be postmenopausal, either following bilateral oophorectomy or at least 5 years after spontaneous menopause. Patients within 5 years of spontaneous menopause or who have had a hysterectomy without bilateral oophorectomy must have postmenopausal LH and FSH levels. Patients on HRT must agree to withdrawal of hormone therapy before letrozole is started. See study protocol for full inclusion/exclusion criteria LOGS Pharmacy Initiation Slides v1.0 25_03_15
Key Exclusion Criteria (1) Patients will be excluded from the study in the following circumstances: • Patients who have had chemotherapy or radiotherapy within 4 weeks (6 weeks for nitrosoureas or mitomycin C) prior to entering the study or those who have not recovered from adverse events due to agents administered more than 4 weeks earlier. • Use of other investigational drugs within 28 days (or five half-lives, whichever is shorter; with a minimum of 14 days from the last dose) preceding the first dose of trametinib or standard of care agent. • Patients may not have received prior MEK, KRAS, or BRAF inhibitor therapy • Current use of a prohibited medication. The following medications or non-drug therapies are prohibited: - Patients may not be receiving any other anti-cancer or investigational agents. - Because the composition, PK, and metabolism of many herbal supplements are unknown, the concurrent use of all herbal supplements is prohibited during the study (including, but not limited to St. John’s Wort, kava, ephedra [ma huang], gingko biloba, dehydroepiandrosterone [DHEA], yohimbe, saw palmetto, or ginseng). See study protocol for full inclusion/exclusion criteria LOGS Pharmacy Initiation Slides v1.0 25_03_15
Key Exclusion Criteria (2) • Patients with known leptomeningeal or brain metastases or spinal cord compression. • Patients with a bowel obstruction or any other gastrointestinal condition that might affect absorption of the oral drug should be excluded. • Patients with a history of interstitial lung disease or pneumonitis. • Patients with a previous or current malignancy at other sites should be excluded, with the exception of: a.) Curatively treated local tumours such as carcinoma-in-situ of the cervix, basal or squamous cell carcinoma of the skin. b.)Tumours for which no relapse has been observed within 5 years • Patient with known Hepatitis B Virus (HBV), or Hepatitis C Virus (HCV) infection (patients with chronic or cleared HBV and HCV infection are eligible). Patients with Human Immunodeficiency Virus (HIV) are not eligible if on anti-retroviral medications. • Known immediate or delayed hypersensitivity reaction or idiosyncrasy to drugs chemically related to Trametinib, or excipients, or to dimethylsulfoxide (DMSO), or to Cremophor EL (polyoxyethylated castor oil). Please note, exclusion for Cremophor is unnecessary unless paclitaxel is the only agent available and the patient randomizes to the conventional therapy option. See study protocol for full inclusion/exclusion criteria LOGS Pharmacy Initiation Slides v1.0 25_03_15
Key Exclusion Criteria (3) • Patients with a history or evidence of cardiovascular risk • Patients with a history or current evidence/risk of retinal vein occlusion . • Any patients with a serious and/or unstable pre-existing medical disorder (aside from malignancy exception above), psychiatric disorder, or other conditions that could interfere with subject’s safety, obtaining informed consent or compliance to the study procedures. • Patients who require use of a concomitant medication that can prolong the QT interval. See the table in section 6.28 of protocol • Pregnant or lactating women. Women of childbearing potential should be advised to avoid pregnancy. See study protocol for full inclusion/exclusion criteria LOGS Pharmacy Initiation Slides v1.0 25_03_15
Treatment Modifications Doses will be reduced for haematological and other adverse events. Dose adjustments are to be made according to the greatest degree of toxicity. Adverse events will be graded using NCI CTCAE v4.0 Control Treatments (Arm A) Dose adjustments as per standard care Trametinib (Arm B) The severity of adverse events will be graded using NCI CTCAE v4.0. Detailed guidelines for dose modifications and interruptions for management of common toxicities associated with the study treatment are provided in section 6.2 of the study protocol. The guidelines outline the dose adjustments for several toxic effects. If a patient experiences several adverse events and there are conflicting recommendations, use the recommended dose adjustment that reduces the dose to the lowest level. The table below outlines the dose levels to be used for any necessary trametinib dose modifications: (Please note: QD = ONCE Daily) A maximum of two trametinib dose level reductions are allowed. If a 3rd dose level reduction is required, treatment will be permanently discontinued. **Please refer to section 6.0 of the study protocol for full details of treatment modifications/dose reductions/delays 15 LOGS Pharmacy Initiation Slides v1.0 25_03_15
LOGS IMP Presentation and Management LOGS Pharmacy Initiation Slides v1.0 25_03_15
General Pharmacy Information (1) The investigational medicinal products in this study are: - Letrozole - Tamoxifen - Paclitaxel - Pegylated Liposomal Doxorubicin - Topotecan - Trametinib (GSK1120212) – experimental agent • All the IMPs for use in the trial with the exception of Trametinib (GSK1120212) will be from sites own stock. There is no provision for funding, reimbursement or discounted stock. • Trametinib will be provided free of charge by GlaxoSmithKline(GSK) and supplied and distributed by Catalent to UK sites for use in the study. • The CRUK CTU, Glasgow will trigger the initial supply of Trametinib for the UK sites at the time of site activation. Delivery will take approximately 5 working days. • Details for re-supply ordering of Trametinib can be found in the IMP Management Document for the study . 17 LOGS Pharmacy Initiation Slides v1.0 25_03_15
General Pharmacy Information (2) • Although specific formulations are mentioned in the study protocol, UK sites are permitted to use locally approved formulations. This must be confirmed to the CR-UK Clinical Trials Unit during initiation process. • Chemotherapy doses may be recalculated every cycle during treatment if it is local practice to do so(e.g. automatic updates by electronic prescribing systems). Where it is not local practice to recalculate every cycle the doses MUST be recalculated if the subject’s weight changes by greater than or equal to 10% from baseline. • BSA calculations should be performed as routine local practice and capped at 2.0m2 • Chemotherapy doses may be dose banded if it is routine local practice to do so. This must be confirmed to the CR-UK Clinical Trials Unit during initiation process. LOGS Pharmacy Initiation Slides v1.0 25_03_15
Prescribing and Dispensing Arrangements • Study specific prescriptions must be used – a master copy must be placed in the pharmacy file • Prescriptions must • Clearly identify prescribing as part of the LOGS study including protocol • Patients study number • Sites are required to include the following information when labelling dispensed supplies for this study: • LOGS Study • Principal Investigator • Eudract Number • Sponsor: NHS Greater Glasgow and Clyde and University of Glasgow • For Clinical Trials Use Only • Patient Trial Number: xxxx • Cycle No: xxxx(if this is local practice to do so) (xxxx – to be completed locally as appropriate There is no stipulation on the format or layout of the labels. Any additional labelling on dispensing can be added as per local practice. LOGS Pharmacy Initiation Slides v1.0 25_03_15
Formulation and Presentation of IMP • Trametinib (GSK 1120212) tablets are immediate release tablets for oral administration. Tablets will be supplied to sites in high density polyethylene (HDPE) bottles that contain a desiccant with a child resistant closure that includes an induction seal liner. • Trametinib (GSK120212) 2mg are pink, round tablets and Trametinib (GSK1120212) 0.5mg are yellow, modified oval tablets. Each pack will contain 32 tablets. All supplies for the LOGS study will be labelled as study specific clinical trial stock. • Trametinib tablets should be stored in a secure area within a refrigerator at a temperature of 2oC – 8oC • All other IMP will be stored and handled as per SmPC LOGS Pharmacy Initiation Slides v1.0 25_03_15
LOGS site file and documentation LOGS Pharmacy Initiation Slides v1.0 25_03_15
Pharmacy Site File • All pharmacy sites will be provided with a pharmacy file containing key documentation and initiation training slides. The pharmacy file must be kept up-to-date and be available for inspection by the study monitors at monitoring visits, regulatory authorities and on request from the sponsor if required. • PSF will include the following: • LOGS IMP Accountability for Trametinib 0.5mg tabs • LOGS IMP Accountability for Trametinib 2mg tabs • LOGS IMP Emergency Supply Log • LOGS Study Specific Training Record • LOGS Study Identification Log • LOGS Study Patient Subject Accountability Log: Arm A (IV) • LOGS Study Patient Subject Accountability Log: Arm A (orals) • LOGS Supply and Receipt Form • LOGS Temperature Deviation & Defect Form LOGS Pharmacy Initiation Slides v1.0 25_03_15
Accountability Logs • Logs must be kept up to date at time of each dispensing and made available if requested for remote monitoring. • Logs can be provided by CRUK CTC for use in this study but local documentation can be used only after approval by CRUK CTC and R&D Sponsor Pharmacy Team. LOGS Pharmacy Initiation Slides v1.0 25_03_15
IMP Accountability (Control Arm A - IV) • Each patient taking part in the study must have a patient log detailing the following information for traceability purposes: – IMP supplied – Date of Issue • Cycle – Dose – Manufacturer, Batch number & expiry date of the product supplied • Diluent batch number & expiry date (where applicable) • Vehicle, batch number & expiry date • Dose banded manufacturer, batch number & expiry date (where applicable) • Aseptic worksheets should be retained • Dose banded is permitted LOGS Pharmacy Initiation Slides v1.0 25_03_15
IMP Accountability Log (Control Arm A- Orals) • Each patient taking part in the study must have a patient log detailing the following information for traceability purposes: – IMP supplied – Date of Issue • Cycle – Dose and frequency • Quantity – Manufacturer, Batch number & expiry date of the product supplied LOGS Pharmacy Initiation Slides v1.0 25_03_15
IMP Accountability (Experimental Arm B- Trametinib)A single Bulk/ Patient IMP accountability log for each strength of trametinib for patients taking part in Arm B must be used detailing the following information: • SHIPMENT • Date • Invoice number • Batch number and expiry date • Quantity received • DISPENSING • Patient study ID and Initials • Quantity dispensed • Batch No and expiry date • Dispensed by/checked by • PATIENT RETURNS • Date • Quantity returned • Received by • PATIENT DESTRUCTION/BULK IMP DESTRUCTION • Date destroyed • Quantity • Signature • Balance remaining on site LOGS Pharmacy Initiation Slides v1.0 25_03_15
Returns and Destruction Patient Returns: • Tablet counts for Control Arm A (orals) must be performed and recorded on the Patient specific Subject Accountability Log. • Tablet counts for Experimental Arm B must be performed on IMP accountability log for Trametinib 0.5mg and 2mg tabs. This is a single log that incorporates both patient and bulk supplies. A separate log should be used for each strength. Destructions: • Patient returns can be destroyed once all accountability has been completed and any discrepancies resolved. • Sites must request permission from sponsor to destroy any expired, damaged or unused stock. LOGS Pharmacy Initiation Slides v1.0 25_03_15
Defects and Temperature Deviations • Complaints or Defects regarding Trametinib: • Complete a copy of the LOGS IMP Temperature Deviation & Defect Form and forward to CRUK CTC and R&D Pharmacy Team. • Complaints or Defects regarding all other IMP: • Should be dealt with by following local hospital procedures. • Temperature deviations regarding Trametinib: • Complete the LOGS Temperature Deviation & Defect Form and forward to CRUK CTC for providing the following information: • Duration of temperature deviation: please provide the maximum period of time the IMP may have been exposed to temperatures out with those indicated • Maximum/ minimum temperature achieved • Quantity of packs and batch number of affected stock • Reason for temperature excursion/any action already taken • Wherever possible please include a copy of the temperature log • Temperature deviation regarding all other IMP: • follow local department procedure but must be notified to the CTU. Further advice will be given as appropriate on a case by case basis. LOGS Pharmacy Initiation Slides v1.0 25_03_15
LOGS Pharmacy Site Initiation Process LOGS Pharmacy Initiation Slides v1.0 25_03_15
Site Set-up CTU GLASGOW Main REC approval - MHRA approval - Site Initiation Slides - Investigator File - Pharmacy File - Sample Collection Supplies SITE Staff Contact & Responsibilities Sheets – SSI - R&D Approval - Investigator CVs and Lead Pharmacist - Delegation log - Clinical Trial Agreement - GCP Certificates for PIs - PIS, Consent, GP Letter etc on Trust headed paper - Lab normal ranges (Haem + Biochem), Accreditation certificates. INITIATION PROCESS DRUG SUPPLY SITE ACTIVATED LOGS Pharmacy Initiation Slides v1.0 25_03_15
Pharmacy Initiation Process • Site initiation process - Each member of the study team is required to participate in site initiation to ensure compliance with the protocol and training on study procedures. Initiation for the study will be done by site staff accessing on line initiation slides via CRUK CTU website • Lead pharmacist for the LOGS study will complete a Pharmacy Site Assessment Form and return to CRUK CTU • A Staff Contact and Responsibilities Sheet must be completed for the lead pharmacist and any other pharmacy clinical trial staff who are delegated IMP management responsibilities. These staff will be required to provided evidence of GCP training and current CV’s • Acknowledgement sheet- Each member of the study who has viewed the initiation slide presentation requires to complete an acknowledgement sheet to confirm this. • Initiation Accreditation call - Prior to activation of the site a short initiation call will be completed with the main contact for the site. LOGS Pharmacy Initiation Slides v1.0 25_03_15
Post Approval • Site Responsibilities • Ensure Pharmacy Site File contents are kept up to date • Ensure accountability logs are kept up to date • Inform CRUK CTU Glasgow of any changes in contacts or arrangements for pharmacy • Action amendments where required. • Sponsor Responsibilities • Forward amendments in a timely manner • Review and amend IMP management process as required • Help solve problems & provide support as required LOGS Pharmacy Initiation Slides v1.0 25_03_15
Contact Details for CR-UK CTU, Glasgow CR-UK CTU, Glasgow Cancer Research UK Clinical Trials Office Level 0, Beatson West of Scotland Cancer Centre 1053 Great Western Road, Glasgow, G12 0YN Tel: +44(0) 141 301 7197 Fax: +44(0) 141 301 7946 E-mail: karen.carty@glasgow.ac.uk (project manager) E-mail: diann.taggart@glasgow.ac.uk (trial co-ordinater) LOGS Pharmacy Initiation Slides v1.0 25_03_15