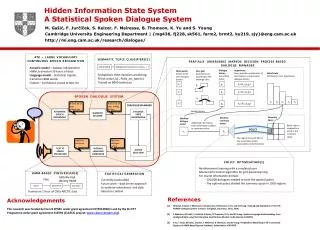
Download

1 / 15
150 likes | 304 Views
system . surroundings . universe . thermodynamic state of a system . The number of moles and identity of each substance. The physical states of each substance. The temperature of the system. The pressure of the system. Thermochemical standard state conditions

E N D
system. surroundings. universe. thermodynamic state of a system. • The number of moles and identity of each substance. • The physical states of each substance. • The temperature of the system. • The pressure of the system. Thermochemical standard state conditions • The thermochemical standard T = 298.15 K. • The thermochemical standard P = 1.0000 atm. • Be careful not to confuse these values with STP. Thermochemical standard states of matter • For pure substances in their liquid or solid phase the standard state is the pure liquid or solid. • For gases the standard state is the gas at 1.00 atm of pressure. For gaseous mixtures the partial pressure must be 1.00 atm. • For aqueous solutions the standard state is 1.00 M concentration. State Functions are independent of pathway: • T (temperature), P (pressure), V (volume), E (change in energy), H (change in enthalpy – the transfer of heat), and S (entropy) Examples of non-state functions are: • n (moles), q (heat), w (work)
There are two basic ideas of importance for thermodynamic systems Law of Conservation of mass and the Law of Conservation of Energy. The second leads to 3 thermodynamic laws. • First Law – the energy of the Universe is constant Chemical systems tend toward a state of minimum potential energy. The first law is also known as the Law of Conservation of Energy. Energy is neither created nor destroyed in chemical reactions and physical changes. Esys +Esurr = EuniverseEsys = KEsys + PEsys • KE – kinetic energy: translational, rotational, vibrational • PE – energy stored in bonds • The second law of thermodynamics states, “In spontaneous changes the universe tends towards a state of greater disorder.” Chemical systems tend toward a state of maximum disorder. The entropy of universe must increase. • Fundamentally, the system must be capable of doing useful work on surroundings for a spontaneous process to occur. In general for a substance in its three states of matter: • Sgas> Sliquid > Ssolid • When: • S > 0 disorder increases (which favors spontaneity). • S < 0 disorder decreases (does not favor spontaneity). • The Third Law of Thermodynamics states, “The entropy of a pure, perfect, crystalline solid at 0 K is zero.”
heat transfer in (endothermic), +q heat transfer out (exothermic), -q w transfer out (-w) Expansion of system w transfer in (+w) Compression of system DG = DH-TDS at constant T and P • The relationship describes the spontaneity of a system. • The relationship is a new state function, G, the Gibbs Free Energy. • Sign conventions for G. • G > 0 reaction is nonspontaneous • G = 0 system is at equilibrium • G < 0 reaction is spontaneous SYSTEM ∆E = q + w
Free energy has the relationship • G = H -TS. • Because 0 ≤ H ≥ 0 and 0 ≤ S ≥ 0, • there are four possibilities for G. • Forward reaction • HSG spontaneity • < 0 > 0 < 0 at all T’s. • < 0 < 0 T dependent at low T’s. > 0 > 0 T dependent at high T’s. > 0 < 0 > 0 Nonspontaneous at all T’s.
Molality is a concentration unit based on the number of moles of solute per kilogram of solvent. Mole fraction • Colligative properties are properties of solutions that depend solely on the number of particles dissolved in the solution. • Colligative properties do not depend on the kinds of particles dissolved. • Colligative properties are a physical property of solutions. • There are four common types of colligative properties: • Vapor pressure lowering • Freezing point depression • Boiling point elevation • Osmotic pressure • Vapor pressure lowering is the key to all four of the colligative properties.
Addition of a nonvolatile solute to a solution lowers the vapor pressure of the solution. • The effect is simply due to fewer solvent molecules at the solution’s surface. • The solute molecules occupy some of the spaces that would normally be occupied by solvent. • Raoult’s Law models this effect in ideal solutions. Boiling Point Elevation Fractional Distillation Freezing Point Depression Osmotic Pressure
Kinetics Four factors that affect the rate of reaction nature of reactant concentration temperature presence of a catalyst 0 order [A] vs t 1/[A] vs t 2nd order ln [A] vs t 1st order Rate of reaction Rate constant Order of reactant Overall order of reaction 1/[A] ln [A] t t General rate expression Initial rate Instantaneous rate Average rate Integrated rate laws ([ ] with respect to time) If zero order [A]0 - [A] = ak t first order second order • Differential rate laws ([ ]with respect to rate) • Rate = k [A]m[B]n[C]p • If zero order Rate = k[A]0 = k • first order Rate = k[A]1 = k[A] • second order Rate = k[A]2 • Change in rate is independent of the change in concentration of a zero order reactant • Change is rate is directly proportional to the change in concentration of a 1st order reactant • Change in rate is directly proportional to the square of the change in concentration of a 2nd order reactant [A] t
Equilibrium Equilibrium is a macroscopic event, regardless of the direction of approach the forward rate and the reverse rate are the same and changes in concentration cannot be observed. Products favored 103 > K > 10-3 Reactants favored Heterogeneous Solutions For mixtures the pure solids, pure liquids, and components that represent large amounts of solvents can be removed from the equilibrium expression and rolled into the equilibrium constant. Mathematical Relationships Reverse the reaction equation K’ = 1/K Scale the reaction equation K’’ = Kn Add equations Krxn = K1∙ K2 ∙ K3 ... Relationship to Kp = K(RT)Dn [C]c[D]d Kc = Predicting reaction direction from non-equilibrium conditions – Q The equilibrium expression for Q is the same as for K. Compare the values of Q and K (the ratios of products to reactants) Q=K the system is at equilibrium Q>K favors reactants Q<K favors products [A]a[B]b To solve for concentrations using K you will need a balanced reaction equation and an ICE table. Factors that affect Equilibrium – [ ], P&V, T For [ ] check Q or use the teeter totter, For gases – P&V, [ ] is to 1/V which is to P, ↑P, ↓V, or ↑[ ] will push to fewer moles of gas. For T check DH is q product or reactant
Buffer Common Ion Effect Titrant Equivalence Point Primary Standard End Point Secondary Standard Titration Solubility Molar solubility Ksp - Solubility Product Constant Ka – Acid dissociation constant Kb – Base dissociation constant Kw – water dissociation constant Le Chatelier’s Principle Lewis Acid Lewis Base Bronsted-Lowry Acid Bronsted-Lowry Base Arrenhius Acid Arrhenius Base Henderson-Hasselbach equation Conjugate acid Conjugate Base pH = -log[H+] pOH = -log[OH-] pKa = -logKa etc. Kw = 1E-14 = [H+][OH-] = 1E-7∙1E-7 pKw = pH + POH = 14 = 7 + 7 Henderson-Hasselbach equation pH = pKa + log([salt]) – log([acid]) pOH = pKb + log([salt]) – log([base]) Determination of strength – or who wins the tug of war for the proton Strong acids Ka > 1 Weak acids Ka < 1 Strong bases Kb > 1 Weak bases Kb < 1
Titration 1. Add solution from the buret. 2. Reagent (base) reacts with compound (acid) in solution in the flask. 3. Indicator shows when exact stoichiometric reaction has occurred. 4. Net ionic equation H+ + OH- --> H2O 5. At equivalence point moles H+ = moles OH-
In General How to Solve Acid/Base Equilibria and Buffer Problems 1. Are the compounds involved strong or weak electrolytes a. If strong then there is a 100% dissociation and no way back use a single headed arrow and when adding the simultaneous equations together only and add the products. i.e. NaOH → Na+ + OH- b. if a weak electrolyte use a double headed arrow and when adding simultaneous equations together add both reactants and products i.e. CH3COOH ↔ CH3COO- + H+ 2. Determine if reactions are neutralization, dissociation, or equilibrium reactions I. Neutralization a. Class I – strong electrolyte/strong electrolyte: produces salt and water. Use ICE table with units of amount (mmol or moles), cancel terms, determine amounts of products. Stop. b. Class II – weak electrolyte/weak electrolyte: Compare Kas or Kbs to determine which predominates products or reactants. Use the equlibrium equation and ICE table with units of concentration (mmol/mL or mol/L) to determine equilibrium concentrations. Stop. c. Class III – strong electrolyte/weak electrolyte: produces salt of the conjugate and water and further actions, Dissociation or Equilibrium reactions must be determined continue to 2 II II. Dissociate any products and begin again at repeat step 1 then continue to 2 III. Products of the dissociation though by name appear to be the same compounds they are not identical in that they have a different source and are treated separately.
III. Equilibrium – write new stoichiometrically balanced chemical equilibrium equations. • Write the equilibrium equation and solve for x and determine [H+] or [OH-] • Find pH or pOH • Determine the pOH from pH or pH from pOH • Determine the [H+] or [OH-] not found in step 3 Ksp Write a stoichiometrically balanced equation for the limited dissociation of the solid If provided with the Ksp and looking for molar solubility of products Write the stoichiometric ratio, i.e. 1:2 Multiply the ratio through by x, i.e. x:2x Write the equilibrium equation, i.e. Ksp = [M2+][Y-]2 Substitute in the stoichiometrically determined x values for the [ ]s, Ksp = (x)(2x)2 Solve for x If provided a mass determined to be in solution and looking for Ksp convert mass to moles convert moles to molarity Write the equilibrium equation, i.e. Ksp = [M2+][Y-]2 Solve for Ksp
Electrochem • The cathode is the electrode at which reduction occurs. • The cathode is positive in voltaic cells. • The anode is the electrode at which oxidation occurs. • The anode negative in voltaic cells. • 1 amp = 1 coulomb/second • In all voltaic cells, electrons flow spontaneously from the negative electrode (anode) to the positive electrode (cathode). • 1. Choose the appropriate half-reactions from a table of standard reduction potentials. • 2. Write the equation for the half-reaction with the more positive E0 value first, along with its E0 value. • 3. Write the equation for the other half-reaction by reverse the tabulated reduction half-reaction and change the sign of the tabulated E0 . • 4. Balance the electron transfer. Do not multiply the potentials. • 5. Add the reduction and oxidation half-reactions. • 6. Add their potentials. • This creates an E0cell which is positive, which indicates that the forward reaction is spontaneous. The Nernst Equation The Gibbs Free Energy Relationship
Primary Voltaic Cells • As a voltaic cell discharges, its chemicals are consumed. • Once the chemicals are consumed, further chemical action is impossible. • The electrodes and electrolytes cannot be regenerated by reversing current flow through cell. • These cells are not rechargable. Secondary Voltaic Cells • Secondary cells are reversible, rechargeable. • The electrodes in a secondary cell can be regenerated by the addition of electricity. • These cells can be switched from voltaic to electrolytic cells. • One example of a secondary voltaic cell is the lead storage or car battery. The Hydrogen-Oxygen Fuel Cell • Fuel cells are batteries that must have their reactants continuously supplied in the presence of appropriate catalysts. • Hydrogen is oxidized at the anode. • Oxygen is reduced at the cathode. • Fuel cells are very efficient. • Energy conversion rates of 60-70% are common!