Download

1 / 15
150 likes | 348 Views
A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Burkhardt , M. F., Martinez, F. J., et al., ( 2013) A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci . 56, 355–364.

E N D
A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells Burkhardt, M. F., Martinez, F. J., et al., (2013) A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 56, 355–364.
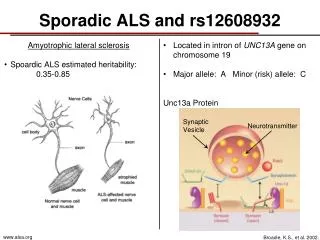
Amyotrophic Lateral Sclerosis (ALS)- genetically complex and progressive neurodegenerative disease • Also known as Lou Gehrig's Disease • Categorized by degeneration of motor neurons (MN) • Affects nerve cells in brain and spine • Degeneration of voluntary functions Introduction
10% of ALS cases are familial (fALS) and 90% are sporadic (sALS) • Pasinelli, P., Brown, R.H.,2006. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710-723. • All ALS patients differ in site of onset, rate of progression, presence of dysfunction, and comorbidity. • Lomen-Hoerth, C., 2011. Clinical Phenomenology and Neuroimaging Correlates in ALS- FTD. J. Mol. Neurosci. 45, 656-662. Review of Literature
The most common and distinct pathology that has been observed across all sALS patients is a change in TAR DNA- binding protein 43 (TDP-43) expression and localization. • Arai, T. et al., 2006. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602-611. • Studies on post mortem tissue has shown that TDP-43 pathology is present in patients with other neurodegenerative diseases (ie. FTLD, Lewy Body Dementia, Alzheimers). • Amador- Ortiz et al., 2007. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 61, 435-445. Review of Literature
The purpose of the study is • To reprogram ALS fibroblasts into iPSCs and differentiate them in order to create a cellular model of sALS through observation Statement of Purpose
Examine different fibroblasts from healthy, fALS, and sALS patients • Overall wanted to create a sALS cellular model • 34 patients • 10 healthy • 8 fALS • 16 sALS Methodology
Custom image analysis Methodology • Flow cytometry • Calcium imaging • Electrophysiology • Screening of Compounds
Data/Results TDP-43 localization in patient iPSC-derived motor neurons.
ALS is a complicated disease • Many different clinical symptoms • Cytoplasmic and intranuclear TDP-43 aggregates • Reported in FTLD patients, fALS patients, and sALS patients • Mislocalization to cytoplasm- increased neuronal death • Mislocalization to nucleus- nuclear dysfunction Discussion
Differences in genetic background and/or interaction with other mutations contribute to heterogeneity of symptoms • Three patients- showed spontaneous TDP-43 pathology • sALS patient population with intranuclear aggregation • Only present in a subset of motor neurons • Generally present in post mortem spinal cord tissue Discussion
Suggests that patients have unidentified genetic lesions that are unapparent as of now • Epigenetic memory related to ALS pathology may have been lost during reprogramming • Focused efforts on sALS • Cause of aggregation is unknown • This model allows for drugs to be able to target specific areas of the nervous system for treatment Conclusion
What can be done in future trials to prevent epigenetic memory loss in reprogramming? • How were the non-aggregate fibroblasts/ iPSCs used if they weren’t found to have aggregates? Questions for the Author