Download

1 / 26
260 likes | 655 Views
Automated NMR Protein Structure Calculation. Peter Guntert RIKEN Genomic Sciences Center, 1-7-22 Suehiro, Tsurumi, Yokohama 230-0045, Japan Accepted 23 June 2003 Παρουσίαση: Βεντούρα Σταυρούλα : std00022@di.uoa.gr. NMR = Nuclear Magnetic Resonance

E N D
Automated NMR Protein Structure Calculation Peter Guntert RIKEN Genomic Sciences Center, 1-7-22 Suehiro, Tsurumi, Yokohama 230-0045, Japan Accepted 23 June 2003 Παρουσίαση: Βεντούρα Σταυρούλα: std00022@di.uoa.gr
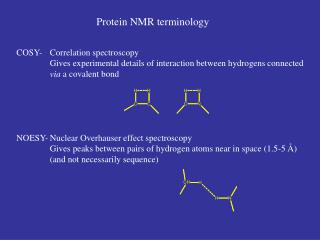
NMR = Nuclear Magnetic Resonance Physical phenomenon based upon the magnetic property of an atom’s nucleus. Studies a magnetic nucleus, by aligning it with an external magnetic field and perturbing this alignment using an electromagnetic field.The response to the field (the perturbing), is what is exploited in NMR spectroscopy. Its used in: 3-dimensional structure of macromolecules Structural genomics Kinetic reactions and properties of proteins What is NMR?
Generally • The transfer of spin polarization from one spin population to another is generally called the Overhauser effect. • in 1950 from American physicist Albert Overhauser • Overhauser effect occurs under different conditions (e.g between electrons and atomic nuclei) yet is most commonly observed and used amongst atomic nuclei and then named Nuclear Overhauser Effect (NOE) • A very common application isNOESY(Nuclear Overhauser Effect SpectroscopY), a magnetic resonancetechnique for structure determination of macromolecular motifs.
This paper • Tries to automate the procedure of determining a 3d-structure of a protein • Chemical – shift assignment • NOESY – assignment, where a refinement of the chemical – shift assignment takes place. • It concentrates on NOESY – assignment, and how it can be automated, with or without the previous step of the chemical-shift assignment.
General principles of automated NOESY assignment and structure calculation (1) Chemical shift assignment Chemical shiftis caused by slight variations in the precession frequency of nuclei due to bonding . This is especially useful in chemical analysis because the precession frequency of each different bonding environment can easily be separated. It is usually expressed in ppm (parts per million) by frequency, and its calculated from
General principles of automated NOESY assignment and structure calculation (2.1) The ambiguity of chemical shift-based NOESY assignment Limited accuracy of chemical shifts values and peak positions many NOESY cross peaks cannot be attributes to a single unique spin pair
General principles of automated NOESY assignment and structure calculation (2.2) A helping model to the problem • Protein with n hydrogen atoms • Correct chemical assignments for these atoms are available • N cross peaks picked in 2D [H-H]-NOESY spectrum with an accuracy of the peak position of Δω • Uniform distribution of the proton chemical shifts over a range of ΔΩ • The chemical shift of a given proton falls within an interval of half-width Δω about a given peak position with probability p = 2 Δω/ ΔΩ.
General principles of automated NOESY assignment and structure calculation (2.4) If 3-dimensional protein structure is known, it can be used to resolve ambiguous NOE assignments. How? If one out of all chemical shift-based assignments possibilities corresponds to a inter-atomic distanceshorter than maximal NOE observable distance dmax. Assuming that the hydrogen atoms are evenly distributed within a sphere of a radius R that represents the protein, the probability q that two given hydrogen atoms are closer to each other than dmax can be estimated by the ratio between the volumes of the two spheres with radii dmax and R. e.g for toxin WmKT : dmax = 5 Å , R = 15 Å then q 4% error
General principles of automated NOESY assignment and structure calculation (3) Automated versus manual NOESY assignment Obstacles: • Cross peaks not sufficient to define the fold of the protein • Erroneously picked picks or inaccurately positioned picks and incompleteness of chemical shift assignments
Algorithms for automated NOESY assignment • Semi-automatic methods • The ASNO method • The SANE method • The NOAH method • The ARIA method • The AutoStructure method • The KNOWNOE method • The CANDID method
The Semi-automatic methodsin general They use the chemical shifts and a model orpreliminary structure to provide the spectroscopist with a list of possible assignments for each cross peak. The user decides interactively about the assignment and/or temporary removal of individual NOESY cross peaks and performs a structure calculation with the resulting,usually incomplete input
ASNO For each cross peak: determination of the set of all possible chemical-shift-based assignments These are checked against the corresponding H-H distances in the available group of preliminary conformers and retained only if the distance between the two protons is shorter than dmax. After several rounds of structure calculation, NOE-assignment and interactive checking and refinement of the assignments, ahigh-quality structure is obtained SANE In SANE (Structure Assisted NOE Evaluation) ambiguous distance constraints are generated for cross peaks with multiple possible assignments. includes a distance filter e.g X -ray structure Minimizes the problem of multiple assignments by using filters. Semi –automatic methodsASNO & SANE
The NOAH method • Programs DIANA and DYANA • Temporarily ignores cross peaks with too many assignments possibilities and generates independent distance constraints for each of the assignment possibilities of the remaining low-ambiguity cross peaks • Initial cycle: all peaks with one or two assignment possibilities are included into the structure calculation. • Large number of erroneous conformational constraints is dealt by attempting to satisfy a max number simultaneously. • Randomly distributed in space, contradicting with correct constraints sets • As a result, they may distort the structure but not lead to a different protein fold. • 70-90% of cross peaks assigned.0.8-2.4% of the assigned had different assignments form manual
The ARIA method (1) • XPLOR and later CNS programs • The use of ambiguous distance constraints. • A NOESY cross peak is treated as the superposition of n degenerate signals from each of its multiple assignments, and interpreted as an ambiguous distance constraint ,b an upper bound on the distance between two H atoms, with
The ARIA algorithm • It starts from lists of peaks and chemical shifts and proceeds in cycles on NOE assignment and structure calculation. • In each cycle: calibrates and assigns the NOESY spectra, merges the constraint lists from different spectra and calculates a bundle of conformers with the program CNS. • Cycle 0 : internally generated extended start structure • Later cycles : NOE assignment, calibration and violation analysis are based on the average distances calculated from the lowest energy conformers from the previous cycle.
The AutoStructure method • AutoStructure program uses rules for assignments similar to those used by an expert to generate an initial protein fold. • Aimsat identifying iteratively self-consistent NOE contact patterns, without using any 3D structure model and delineating secondary structures. • Generates conformational constraints e.g distance, and submits parallel structure calculations with the program DYANA. The remaining structure is then refined automatically by iterative cycles of self-consistent assignment of cross peaks and regeneration of the protein structure with DYANA.
The KNOWNOE method • Knowledge driven Bayesian algorithm for resolving ambiguities in NOE assignments. • Cross peak with n possible assignments A1,…,An • P (Ak ,α|V). Ak is responsible for at least a fraction α of the cross peak volume V. • Higher than a cutoff, considered unambiguously assigned. • Set of structures is calculated with unambiguously assigned peaks. They are used as input for a next cycle in which those assignments are accepted with distances shorter than a threshold dmax (decreased till 5 Å) • Requires: High accuracy chemical shifts of 0.01ppm
The CANDID method (1) • NOAH and ARIA • Network-anchoring • Evaluates the self- consistency of NOE assignments independent of knowledge on the 3D protein structure and in this way compensates for the absence of a de novo structure determination. • Detects erroneously constraints that might artificially constrain unstructured parts of the protein • Constraint combination • Aims at minimizing the impact of imperfections (spurious constraints) on the resulting structure at the expense of temporary loss of information. • Consists of generating distance constraints with combined assignments from different cross peaks.
The CANDID method (3)Overview of the algorithm • Seven cycles. Information transferred through 3D structures. • A cycle starts: generates for each NOESY cross peak an initial assignment list containing the H atoms pairs that could contribute to the peak. These assignments are weighted with criteria and initial assignments of low overall score are discarded. • The output of a cycle:List of cross peaks assignments,of comments about decisions that can help to recognize artifacts in the input data… • First cycle, network anchoring has a dominant impact • Constraint combination used in the first two cycles. • Final cycle: additional filtering for making sure that all NOEs have unique assignments.
Robustness and quality control of automated NMR structure calculation • Effect of incomplete chemical shift assignments • 90% completeness of the chemical shift assignment • Random omission of entries from the lists • From peaks involved in many NOEs • From peaks ‘unimportant’ involved in fewer NOEs • Effect of incomplete NOESY peak picking • Omission up to 50% of the cross peaks • Quality control • Criteria needed to specify if a final structure from an automatic algorithm is ‘correct’.
Structure calculation without chemical shift assignment (1) • General idea: Exploit the fact that NOESY spectra provide distance information even in the absence of any chemical shift assignments. • Protons are treated as a gas of unconnected particles. • First tested 1992 from Malliavin et al. with back-bone protons of lysozyme. • NOEs provide measurements with 5% accuracy. • Absence of a NOE means that distance exceeds 4.5 Å • No chemical shift degeneracy • Algorithms can extract from the proton clouds the assignments of backbone H atoms with less than 10% error
Structure calculation without chemical shift assignment (2) The ANSRS method(’94) • Input: A list of NOESY cross peaks including • Knowledge of chemical shifts of C or N atoms bound to the protons that make the NOE • Complete but unassigned list of the chemical shifts of all H – C or H – N moieties. Algorithm First step : 3D structures of unconnected H atoms are calculated using dynamical simulated annealing Second step : List for each residue type of H spin combinations with probability scores Third step: Sequence-specific assignment and a low-resolution 3d structure are obtained by simulated annealing.
Structure calculation without chemical shift assignment (3) • Atkinson and Saudek • Optimization of four variables per atom, three Cartesian coordinates and the chemical shift value directly from the spectrum • The direct determination of proteins by NMR without chemical shift assignment can incorporate other spectra The Clouds method • Combines the proton cloud model in conjunction with molecular dynamic annealing information.