Download

1 / 29
320 likes | 400 Views
Discover the different methods for analyzing protein structures, including alignment, prediction, and comparison with emphasis on homology modeling and threading. Learn about tools like DALI and VAST for structure alignment and secondary structure prediction. Evaluate prediction performance and examine the significance of protein structure comparison.

E N D
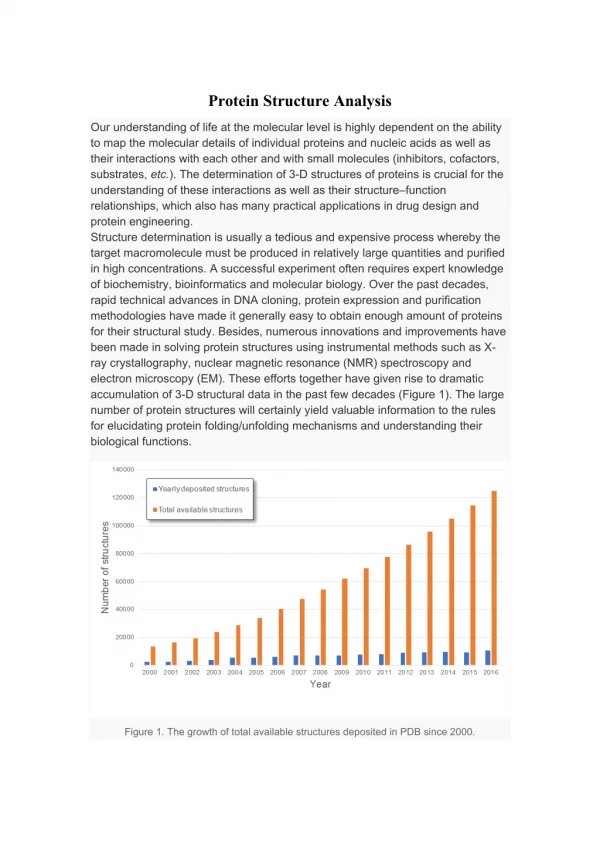
PLPTH 890 Introduction to Genomic Bioinformatics Lecture 23 Protein Structure Analysis - II Liangjiang (LJ) Wang ljwang@ksu.edu April 10, 2005
Outline • Protein structure alignment (DALI and VAST). • Protein secondary structure prediction (PHDsec, PSIPRED, etc). • Prediction of 3-D protein structures: • Homology modeling. • Threading. • Ab initio prediction. • Protein structural genomics.
Protein Structure Comparison • Why is structure comparison important? • To understand structure-function relationship. • To study the evolution of many key proteins (structure is more conserved than sequence). • Comparing 3-D structures is much more difficult than sequence comparison. • Protein structure classification: • SCOP: Structure Classification Of Proteins. • CATH: Class, Architecture, Topology and Homology. • Protein structure alignment: DALI and VAST.
Protein Structure Alignment • Positions of atoms in two or more 3-D protein structures are compared. • Must first determine which atoms to align. At least two sets of three common reference points should be identified. • Atoms in structures are • matched to minimize the • average deviation. • Computers are NOT good • at comparing 3-D objects • (an NP-hard problem). (Baxevanis and Ouellette, 2005)
How to Compare Structures? Structure 1 Structure 2 Feature extraction Description 1 Description 2 Comparison Scores Statistical analysis Similarity, classification
DALI • DALI is for Distance matrix ALIgnment. • Each structure is represented as a two-dimensional array (matrix) of distances between all pairs of C atoms. • Remember what aCatom is? • Assume that similar 3-D structures have similar inter-residue distances. • DALI uses distance matrices to align protein structures. • DALI is available at http://www.ebi.ac.uk/dali/.
VAST • VAST is for Vector Alignment Search Tool. • Each structure is represented as a set of secondary structure elements (SSEs). • SSEs: helices or strands. • VAST scores pairs of SSEs based on their type, orientation and connectivity. • The SSE matches of statistical significance are then extended (similar to BLAST). • Structures in MMDB have been pre-computed, and organized as structure neighbors in Entrez. • VAST can be accessed athttp://www.ncbi.nlm.nih.gov/Structure/VAST/vast.shtml.
Secondary Structure Prediction • Given the sequence of a polypeptide, secondary structures are predicted. • Assume that secondary structures are fully determined by local interactions among neighboring residues. • Early analysis were based on the frequencies of amino acid found in different types of secondary structures. • For example, proline occurs at turns, but not in helices. • Modern approaches use machine learning techniques and multiple sequence alignments.
Machine Learning Approach QEALDAAGDKLVVVDF HHHHHHLLLLEEEEEE H – Helix E – Sheet L – Loop Training Dataset Test Dataset Training Testing Classifier (Model) No Yes Prediction Performance?
Input layer S P A R S K Y Hidden layer Output layer H E L PHDsec • For a given protein sequence: • Search for homologous sequences. • Produce a multiple sequence alignment. • Generate a profile (evolutionary information). • PHDsec uses a feed-forward artificial neural network to predict the secondary structures. (PHDsec can be accessed at http://www.predictprotein.org/)
PSIPRED • For a given protein sequence: • Perform a PSI-BLAST search. • Create a profile that conveys the evolutionary information at each position. • Feed the profile into a system of neural networks (or support vector machines). • PSIPRED can be accessed at http://bioinf.cs.ucl.ac.uk/psipred/.
How to Evaluate the Performance? • EVA: an independent server for evaluation of protein structure prediction methods. • The best tool • for three-state • per-residue • secondary • structure • prediction • now reaches • the accuracy • of about 78%. (http://cubic.bioc.columbia.edu/eva/)
Prediction of 3-D Protein Structures • There are about 30,000 structures in PDB, but more than 1.8 million non-redundant protein sequences in UniProt (Swiss-Prot + TrEMBL). • Computational structure prediction may provide valuable information for most of the protein sequences derived from genome sequencing projects. • Three predictive methods: • Homology (or comparative) modeling. • Threading (or fold recognition). • Ab initio structure prediction.
Sequence-Structure Relationship • In cells, protein folding is determined by the amino acid sequence. But, protein structures can also be affected by post-translational modifications and the cellular environment. • Proteins with ≥ 30% sequence identity tend to have similar structures. However, exceptions do exist … 80-residue stretch (yellow) with 40% sequence identity (Bourne, 2004) (Viral capsid protein, 1PIV:1) (Glycosyltransferase, 1HMP:A)
Homology Modeling • Probably the most accurate method for protein structure prediction. • Five different steps: • Find a known structure related to the query sequence by sequence comparison. • Align the query sequence with the known structure (template). • Build a model by modifying the backbone and side chains of the template. • Refine the model using energy minimization. • Validate the model using visual inspection or software tools.
Homology Modeling (Cont’d) • Accuracy of structure prediction depends on the percent amino acid sequence identity shared between the query and template. • For >50% sequence identity, RMSD (Root Mean Square Deviation) is only 1 Å for main-chain atoms, which is comparable to the accuracy of a medium-resolution NMR structure or a low-resolution X-ray structure. • Homology modeling may not be used for predicting protein structures if the sequence identity is less than 30%.
Homology Modeling Servers • SWISS-MODEL (http://swissmodel.expasy.org/): A popular site for structure homology modeling. • SDSC1 (http://cl.sdsc.edu/hm.html): • the #1 ranked • server for • homology • modeling on • the EVA site. SDSC1 http://cubic.bioc.columbia.edu/eva/
(Baxevanis and Ouellette, 2005) Threading
Threading (Cont’d) • Threading takes a query sequence and passes (threads) it through the 3-D structure of each protein in a fold database (known structures). • As a sequence is threaded, the fit of the sequence in the fold is evaluated using some functions of energy or packing efficiency. • Threading may find a common fold for proteins with essentially no sequence homology. • Structures predicted from threading techniques often are not of high quality (RMSD > 3 Å). • Based on EVA results, 3D-PSSM is the best threading server (http://www.sbg.bio.ic.ac.uk/~3dpssm/).
Ab Initio Structure Prediction • Ab initio prediction can be used when a protein sequence has no detectable homologues in PDB. • Protein folding is modeled based on global free-energy minimization. • Since the protein folding problem has not yet been solved, the ab initio prediction methods are still experimental and can be quite unreliable. • One of the top ab initio prediction methods is called Rosetta, which was found to be able to successfully predict 61% of structures (80 of 131) within 6.0 Å RMSD (Bonneau et al., 2002). • The HMMSTR/Rosetta Server can be accessed athttp://www.bioinfo.rpi.edu/~bystrc/hmmstr/server.php.
Comparing Structure Prediction Methods A – C: homology modeling with 60% (A), 40% (B) and 30% (C) sequence identity. D and E: ab initio protein structure prediction. Predicted structures are in red, and actual structures are in blue. (Baker and Sali, 2000)
Example: Cysteine-Rich Peptides Signal helix and cleavage site C C C C C C C C NCR:Nodule-specific Cysteine Rich genes in legumes. Avr9: fungal avirulence protein from Cladosporium fulvum. Defensin: antimicrobial peptides. Proteinase inhibitor: Serine proteinase inhibitors. SCR6:S-locus of Brassica, SI, interact with SRK6.
Ab Initio Prediction of Cys Rich Peptides LSG-TC51151 PsENOD3 Defensin Avr9 (AAG40321, M. sativa) (Cladosporium fulvum)
Protein Structural Genomics • A worldwide initiative aimed at determining a large number of protein structures in a high throughput mode. • In the US, nine structural genomics centers have been funded by the National Institutes of Health (NIH). • More information may be found at http://www.rcsb.org/pdb/strucgen.html. • TargetDB (http://targetdb.pdb.org/): a centralized registration database for target sequences from the worldwide structural genomics projects.
A Target Selection Pipeline from JCSG Methods TMHMM Protein size (7 - 80 kDa) Low complexity Redundancy BLAST against PDB sequences
Summary • Fast and accurate structure alignment is still a very hard problem to be solved. • Machine learning techniques are widely used in protein secondary structure prediction. • Homology modeling is probably the most reliable method for structure prediction. • The protein folding problem has not yet been solved.
Prediction of Solvent Accessibility • Solvent accessibility: the relative area of a residue’s surface that is exposed to the surrounding solvent. • The solvent-accessible residues may be part of an active site or a binding site, while the buried residues may play an important role in stabilizing the protein structure. • PHDacc (http://www.predictprotein.org/): a neural network-based method (similar to PHDsec). • Jpred (http://www.compbio.dundee.ac.uk/~www-jpred/): a neural network system that predicts both secondary structure and solvent accessibility.
Predicting Transmembrane Segments • Transmembrane segments share common biophysical features (e.g., hydrophobicity). • PHDhtm (http://www.predictprotein.org/): • Part of the PredictProtein services. • Transmembrane helices are predicted using a neural network system. • TMHMM (http://www.cbs.dtu.dk/services/TMHMM/): • A set of known transmembrane segments are represented as HMMs. • A query sequence is matched to a known transmembrane pattern.
Signal Peptide Prediction • Extracellular proteins or proteins targeted to subcellular compartments contain short signal peptides (often at the N-terminal). • PSORT (http://psort.ims.u-tokyo.ac.jp/): A rule-based expert system for predicting subcellular localization of proteins from their amino acid sequences. The algorithm of k-nearest neighbors is used for reasoning. • SignalP (http://www.cbs.dtu.dk/services/SignalP/): predicts the presence and location of signal peptide cleavage sites using a combination of neural networks and HMMs.