Download

1 / 18
180 likes | 299 Views
Supplemental information. Sequence collection qPCR calibration formula. Database subcollections and their taxonomy relationship. Mollusca. Symbol. Database. Gastropoda. Patellogastropoda. Lottia gigantea. Lgi. Genome, EST. Vetigastropoda. Haliotis. Haliotis asinina. Has. EST.

E N D
Supplemental information Sequence collection qPCR calibration formula
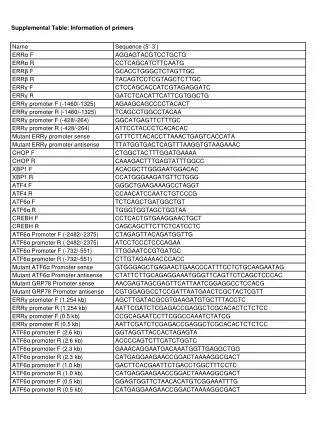
Database subcollections and their taxonomy relationship Mollusca Symbol Database Gastropoda Patellogastropoda Lottia gigantea Lgi Genome, EST Vetigastropoda Haliotis Haliotis asinina Has EST Haliotis midae Hmi Transcriptome Haliotis diversicolor Hdiv Transcriptome Haliotis discus Hdis EST Caenogastropoda Littorina littorea Lli mRNA Heterobranchia Aplysia californica EST Bivalvia Heteroconchia Meretrix meretrix Mme Transcriptome Pteriomorphia Ostreoida Crassostrea angulata Can Transcriptome Mytiloida Mytilus galloprovincialis Mga EST Cephalopoda Octopus vulgaris Transcriptome
SARP19 sequence collection- Step1. AA sequences of H. diversicolor SARP19-I1 (GB: JU063184) and Littorina littorea SARP19 (GB: AAM20842) were BLASTp to GenBank NR protein database. Hits distributed in a wide taxonomy catalog from nematodes to birds (FigS1). Such wide distribution could be resulted from the conservative EF-hand calcium-binding motifs. However, some of the distances are too long to fit monophyletic hypothesis. Sequence collection should be focused.
FigS1. Hitting pattern SARP19-I1 to GenBank NR protein database
SARP19 sequence collection- Step2. • EST or TSA sequence libraries of three abalones, H. diversicolor, H. discus and H. asinina (EST), were downloaded from NCBI. • CDS of H. diversicolor SARP19-I1 and Littorina littorea SARP19 were tBLASTx these mRNA libraries. Similar sequences were identified and repeated such BLAST until no new sequence was added. • Redundant sequences were manually removed, and CDSs were predicted by interpreting the BLAST results. Because some sequencing errors can cause breaks in the CDS, the obvious break points were manually modified by adding or deleting single nucleotides to replace the CDS. • These CDS were BLAST Lottia gigantea genome (http://genome.jgi-psf.org/pages/blast.jsf?db=Lotgi1) and EST. New non-redundant CDS were added to the collection. • Alignments of putative amino acid sequences were performed by ClustalW and then manually modified. Neighbor-joining trees were constructed by MEGA (FigS2).
FigS2. NJ tree of SARP19-like sequences collected from three abalones, Lottia gigantea and Littorina littorea.○, H. diversicolor; ●, Haliotis discus; ■, Haliotis asinina; △, Lottia gigantea; ▲, Littorina littorea
SARP19 sequence collection– Step3. • mRNA libraries of Crassostrea angulata and Meretrix meretrix were added. After search, 12 more SARP19-like sequences were recruited. • The NJ guild tree shows the collection could be separated as two distinct groups (FigS3, S4). • Average branch length is obviously different between Group A and B. It may imply that their evolutionary constraints were different.
Group A FigS3. NJ tree of 27 SARP19-like sequences collected from ○, H. diversicolor; ●, Haliotis discus; ■, Haliotis asinina; △, Lottia gigantea;▲, Littorina littorea ;◇, Crassostrea angulata; ◆, Meretrix meretrix. Group B
FigS4. NJ tree of 27 SARP19-like sequences collected from ○, H. diversicolor; ●, Haliotis discus; ■, Haliotis asinina; △, Lottia gigantea; ▲, Littorina littorea ; ◇, Crassostrea angulata; ◆, Meretrix meretrix. Group A Group B
SARP19 sequence collection– Step4. • More mRNA sequence libraries were added: Haliotis midae (Bioproject: PRJNA79815), Aplysia californica (EST), Octopus vulgaris (Bioproject: PRJNA79361) • After search, 12 more mRNA sequences from H. midae were recruited. No SARP19-like sequence was found from sea hare and octopus neural transcriptoms. • The NJ guild tree was built as previous described (FigS5) .
FigS5. NJ tree of 39 SARP19-like sequences.○, Haliotis diversicolor; ●, H. discus; □ , H. midae; ■, H. asinina; △, Lottia gigantea; ▲, Littorina littorea ; ◇, Crassostrea angulata; ◆, Meretrix meretrix; Group A Outgroup for GroupA Collection boundary Group B
SARP19 sequence collection– Step5. • Setting the collection boundary • Best hitting. All sequences from Group A show best hitting to other members in Group A. However, some sequences of Group B show ambivalent best hitting pattern. • To simplify the situation, a boundary was set as showed in FigS5. Those ambivalent sequences could act as outgroup of Group A. • 26 sequences of the collection were final go-through SWISSPROT, NR and NT databases to find any new sequences that fit the boundary. However, no new sequence was qualified.
Vdg3sequence collection- Step 1. AA sequences of H. diversicolor Vdg3-I1 were BLASTp to GenBank NR protein database. The hitting pattern was much simpler than SARP19 (FigS6). No conserved motif was found in the putative vdg3 proteins.
FigS6. H. diversicolor vdg3-I1 Hitting to GenBank NR protein database
Vdg3sequence collection- Step 2. • CDS of H. diversicolor vdg3-I1 and H. asinina vdg3 were hit to former constructed sequence libraries and Lottia gigantea genome (http://genome.jgi-psf.org/pages/blast.jsf?db=Lotgi1). • Similar sequences were identified and they were repeated such hitting until no new sequence was added. • As former procedure, redundancies were removed, CDS were predicted and patched, alignments and NJ trees were constructed, and collection boundaries were set by best hitting or guild trees. • Sequences of the collection were final go-through SWISSPROT, NR protein database and nt DNA database to recruit any missed sequences that fit the boundary. • The NJ tree of 30 vdg3-like proteins was showed in FigS7.
FigS7. NJ tree of 30 vdg3-like sequences. ○ H. diversicolor; ● H. discus; □ H. midae; ■ H. asinina; △ Lottia gigantea; ▲ Littorina littorea; ◇ Crassostrea angulata; ◆ Meretrix meretrix; ▼ Mytilus galloprovincialis.
qPCR calibration formula • For a set of qPCR reactions in a same run, fluorescence intensities (designate as F) should be constant (set as f) when they reach their threshold cycle numbers (Ct) i.e., FCt = f (1). • While the fluorescence intensity F in a SYBR Green qPCR system is in direct proportion to the DNA amount of an amplicon, then F = k•N•L (2), where k is an unknown constant, N is the molecular number and L is the amplicon length. • While N = N0 • E^Ct (3), where N0 is the initial molecular number and E is the PCR efficiency; then, for each gene k •N0(gene) • E(gene)^Ct(gene)•L(gene)= f (4). • Then, we have the calibration formula N0(target gene) / N0(control gene) = (E(control gene)^Ct(control gene)•L(control gene))/ (E(target gene)^Ct(target gene)•L(target gene)) (5), where OAZ1 was set as control gene and N0(control gene) in each stage was set as 100.