Download
1 / 52
530 likes | 584 Views
Explore the synthesis, structure, and functions of hemoglobin, and learn about its oxygen-binding properties, dissociation curve, testing methods, and related disorders like sickle cell anemia.

E N D
1. Introduction 2. Synthesis and structure of Hemoglobin (Hb) • Synthesis of Heme • Synthesis of globin chains 3. Functions of Hemoglobin 4. Oxygen-Hemoglobin dissociation curve 5. Tests to detect Hemoglobin /blood 6.Hemoglobin Derivatives 7.Hemoglobinopathies • Sickle cell anemia • Thalassemisa
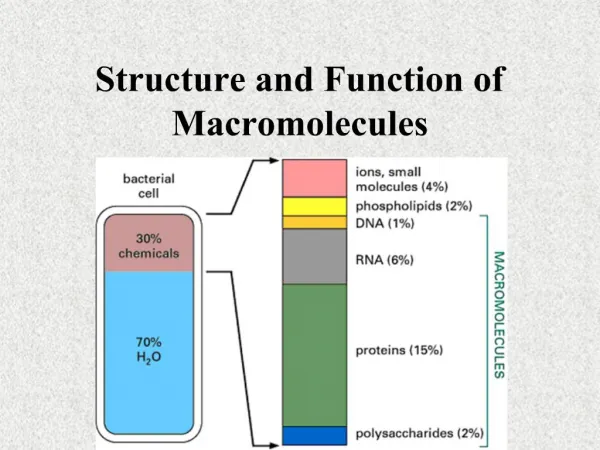
Introduction • The main function of red blood cell • Transfer of O2 from lungs to tissue • Transfer of CO2 from tissue to lungs • To accomplish this function red cells has hemoglobin (Hb) • Each red cell has 640 million molecules of Hb
Introduction • Hemoglobin (Hb), protein constituting 1/3 of the red blood cells • Synthesis begins in proerythroblast • 65% at erythroblast stage • 35% at reticulocyte stage • Two parts • Heme • Globin
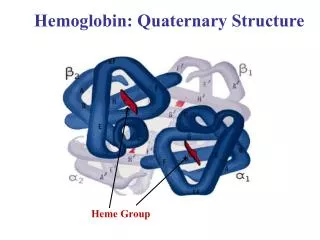
Concentration of hemoglobin in blood in males is 14-16 g/dl and in females , 13- 15 g/dl. • Hemoglobin is globular in shape . • Molecular weight of adult hemoglobin is 67,000 Daltons. • The globin part of Hb is made up of 4 polypeptide chains (i.e., tetramer). Each polypeptide chain has a heme moiety attached to it.
Synthesis of Hemoglobin (Hb) • Heme & globin produced at two different sites in the cells • Heme in mitochondria • Globin in polyribosomes • Well synchronized
Synthesis of Heme • Protoporphyrin ring with an iron atom in centre • The main site is mitochondria as it contains ALAS • Mature red cell does not contain mitochondria
Synthesis of globin • Various types of globin combines with heme to from different hemoglobin • Eight functional globin chains, arranged in two clusters • b- cluster (b, g, d and eglobin genes) on the short arm of chromosome 11 • a- cluster (a and zglobin genes) on the short arm of chromosome 16
Synthesis of globin Globin synthesis, starts at 3rd week of gestation • Embryonic Hemoglobin Gower I ( z2e2) Hemoglobin Portland ( z2g2) Hemoglobin Gower II (a2e2) • Fetal : HbF (a2g2), HbA (a2b2) • Adult : HbA, HbA2 ( a2d2), HbF.
Each Alpha chain has 141 amino acids . • The beta , gamma, delta chains have 146 amino acids. • There are 36 histidine residues in Hb molecule, which are important in buffering action .
Primary Structure of Hemoglobin : The globin chains are of two types . i.e., (i) alpha chain (141 amino acids) - common in all forms of Hb and (ii) beta , gamma, delta ,either , or (146 amino acids). • Secondary structure : The globin chain have helical segments interrupted by non-helical segments, which permit the polypeptide chains to fold into tertiary structure. • Tertiary structure : The polypeptide chains fold upon themselves to form a coiled structure, with a central hydrophobic core that acts as a heme pocket. • Quaternary structure : Globin or the four polypeptide chains or the subunits are linked to one another by ionic bonds
Functions of Hemoglobin 1. Oxygen delivery to the tissues 2. Transport of CO2 3. Buffering Action
1. Oxygen delivery to the tissues • Reaction of Hb & oxygen • Each Hb molecule has 4 heme groups and can bind a maximum of 4 molecules of O2 at a time. • Less than .01 sec required for oxygenation • beta chain move closer when oxygenated • When oxygenated 2,3-BPG is pushed out • beta chains are pulled apart when O2 is unloaded, permitting entry of 2,3-BPG resulting in lower affinity of O2
Oxygen-hemoglobin dissociation curve • The ability of hemoglobin to load and unload oxygen at physiological PO2 (partial pressure of O2) is described by the oxygen dissociation curve (ODC) • Sigmoid shape
In lungs PO2 is high (100 mm Hg). Therefore, hemoglobin gets fully saturated with O2. • In the peripheral tissues, PO2 is low (20 mm Hg) and therefore, oxyhemoglobin releases O2 for cellular respiration. • The oxygen dissociation curve of Hb is sigmoid shaped indicating cooperative binding of O2, i.e. binding of one O2 molecule to one heme increases the affinity of O2 to other heme moieties facilitating uptake of O2. Similarly, release of O2 from one heme decreases the affinity in other heme moieties and facilitates the release of O2. • Cooperative binding of O2 is due to conformational change in the Hb molecule
Conformation of deoxy hemoglobin is said to be in T state (tense/taut state) and that of HbO2 in R state (relaxed state
Hb-oxygen dissociation curve • The normal position of curve depends on • Concentration of 2,3-BPG • H+ ion concentration (pH) • CO2 in red blood cells • Structure of Hb
Hb-oxygen dissociation curve • Right shift (easy oxygen delivery) • High 2,3-DPG • High H+ • High CO2 • HbS • Left shift (give up oxygen less readily) • Low 2,3-DPG • HbF
2. Transport of CO2 • About 200 ml of CO2 is produced per minute in tissues at rest in the following forms i) 75% of CO2 is transported as bicarbonate (HCO3–) • Chloride shift or Hamburger effect..
Isohydric transport of CO2 by Hb Co2+H2o H2Co3 H+ +Hco3
The Bohr Effect Competition between oxygen and H+ Discovered by Christian Bohr Binding of protons diminishes oxygen binding Binding of oxygen diminishes proton binding Important physiological significance
Transport of CO2….. • ii) 15% of CO2 produced is transported as carbamino hemoglobin : • CO2 binds to the free -amino groups at the N-terminal of the globin chains in Hb to form carbamino compounds. • iii) About 10% of CO2 is transported as dissolved form.
3. Buffering Action • Hb transports CO2 from tissues to the lungs as HCO3- with minimum change in pH. • Each Hb molecule binds 2 protons generated from CO2 and H2O. CO2+ H2O H2CO3 H+ +HCO3- HbHHb CA in RBC Peripheral tissues
Tests to detect Hemoglobin /blood • Benzidine test : for blood in urine (Qualitative) • Hemin crystals • Spectroscopy : absorption spectra used for clinical and forensic applications
Hemoglobin Derivatives • Methemoglobin • Carboxyhemoglobin
Methemoglobin • Hb with Fe3+ ions is known as methemoglobin (met-hb). • Formation of met-hb : oxidizing agents such as H2 O2, free radicals or oxidant drugs (such as sulfonamides), etc., May cause this oxidation of Fe2+ to Fe3+, may also be hereditary as in congenital methemoglobinemia.
Biological role of met-hb: met-hb cannot bind oxygen since Fe3+ cannot bind o2. • Normally small quantities ( 1%) of methemoglobin are formed in rbcs which get reduced back readily to fe2+ state by the enzyme methemoglobinreductase.
Clinical relevance of methemoglobin in cyanide poisoning The consequences of cyanide poisoning are • combines with met-Hb in blood • inhibition of cytochromeoxidase in the ETC. • Antidote: Amyl Nitrite is used as an antidote for Cyanide poisoning. Nitrites oxidise the Fe++ iron to Fe+++ in heme forming met-Hb which has a high affinity for cyanide. So cyanide will not be released to the tissue from the blood. Met-Hb formation is of less serious consequence than inhibition of cytochromeoxidase. • Symptoms : Cyanosis, Increased deoxyHb. • Treatment : with ascorbic acid.
Carbon MonoxyHb / Carboxy – Hb (CO-Hb) Carbon monoxide has the ability to bind Hb (same way as oxygen) forming CO-Hb, and Hb has 200 times more affinity for CO than O2. • Normal levels : Normal people have about 0.16% of CO-Hb. An average smoker has an additional 4% of CO-Hb. • Clinical symptoms manifest when CO-Hb levels exceed 20%. • At about 40-60% saturation, coma and death can occur. • Detection of met-Hb and CO-Hb : Both met-Hb and CO-Hb may be detected by spectroscopy by their characteristic absorption bands in the spectrum.
Glycated hemoglobin Non enzymatic post-translational modification by combination of NH2 terminal amino group of Hbβ-chain and glucose. 4 – 6% normal
Hemoglobin Variants : • Over 900 mutant human Hb’s are known. However, they are rare and benign showing no clinical abnormalities.
Hemoglobinopathies When a mutant Hb causes clinical abnormality (loss of biological function), the condition is termed hemoglobinopathy. The genetic defect (mutation) may result in abnormal Hb (i) Synthesis of structurally abnormal globin (altered primary structure of a chain) with altered function. E.g., : Sickle cell anemia (HbS), hemoglobin C disease (Hbc) (ii) Synthesis of globin chains in insufficient quantities. E.g., thalassemia.
Sickle Cell Hemoglobin (HbS) and Sickle Cell Anemia Occurrence : This is the most common form of mutant/abnormal Hb. • Sickle cell anemia is largely confined to tropical areas of the world primarily in black population. Inheritance of HbS : • Mode of inheritance is autosomal recessive, • HOMOZYGOTES: only the homozygotes show clinical symptoms – the condition is referred to as sickle cell anemia or sickle cell disease.Here the erythrocytes get distorted to characteristic sickle shape at low oxygen concentration. • HETEROZYGOTES have sickle cell trait, do not show clinical symptoms and can have a normal life span.
Mechanism of Sickling : Defect in HbS : substitution of glutamate by valine in the 6th position of beta globin chains.
The substitution of this single amino acid generates a hydrophobic, sticky patch, on the outer surface of chains, of both deoxy-HbS and oxy-HbS. • Deoxy HbA has a hydrophobic patch on its surface, which is complimentary to the stick patch of deoxy HbS. The sticky patch of one deoxy HbS binds to the complimentary of another deoxy HbS, thus an aggregate of deoxy HbS molecules is formed. • This polymerization of deoxy HbS molecules leads to the formation of long fibrous precipitates, which distort the RBCs into sickle or crescent shape
Clinical Manifestations of Sickle Cell Anemia : • Clinical manifestations are due to sickled RBC’s which are fragile causing hemolysis, blocking capillaries causing poor blood supply to tissues. This results in 1. Life long hemolytic anemia 2. Tissue damage and pain 3. Increased susceptibility to infections and 4. Premature death.
Heterozygous Individuals are resistant to Falciparum Malaria Malarial parasite (plasmodium falciparum) spends a part of its life cycle in RBCs. Since sickled RBCs have a shorter life span the life cycle of the parasite is affected . Diagnosis : Sickle cell anemia is diagnosed by electrophoresis, where HbS migrates faster than HbA towards anode. • (Each HbS molecule contains additional 2 negative charges of the substituted glutamate residues)
Thalassemias • Thalassemias are a group of hereditary hemolytic disorders characterized by impairment in the synthesis of globin chains. Molecular Basis : • Thalassemias are characterized by a defect in the production of either alfa or beta - globin chain, but there is no abnormality in the primary structure of the individual chains.
There are two types of thalassemias depending on the globin chains that are defective. • Alfa -Thalassemia : This is caused by decreased synthesis or complete absence of Alfa - globin chains. • Beta - Thalassemias :Results due to decreased synthesis or total lack of β- globin chains. • In thalassemia life span of RBCs is decreased leading to haemolytic anemia.