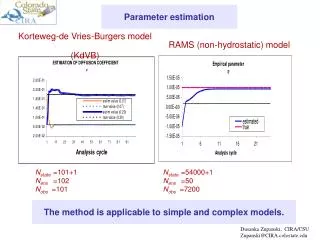
Download

1 / 20
200 likes | 334 Views
Code-Level Parameter Estimation. The Dryest Presentation Ever Bob Zimmermann 7 September 2005. Annotations. Parameters!. Sequences. What is it that We’re Doing Here Again?. An object-oriented, extensible parameter estimator A parameter estimator with minimized redundant code

E N D
Code-Level Parameter Estimation The Dryest Presentation Ever Bob Zimmermann 7 September 2005
Annotations Parameters! Sequences What is it that We’re Doing Here Again? • An object-oriented, extensible parameter estimator • A parameter estimator with minimized redundant code • A usable parameter estimator
Overview Parameter Estimation has 5 main phases: • Instantiation • Read in config files, initialize gHMM • Annotation • Convert annotations to state sequences • Segment the annotations • Regioning • Convert annotations to regions • Counting • Count the models • Estimation
Instantiation:What the User Sees • 3 levels of configuration • Instance file: command line options describing the sequences & annotations to be estimated • gHMM file: HMM description, model description, null region description • Feature Map file: describes the conversion required to get from annotation to state sequences. • User only inputs an instance file
Annotation:Steps • Each annotation is read in one by one, possibly by chromosome • Any number of sequences are associated with each annotation • Annotations are converted into features • Null regions are applied to appropriate features • Segmentation
Einit0 Intron0 Exon0 Einit0 Intron0 Exon0 Annotation:A Review of Layering and Segmentation
Eterm0 Eterm0 Einit0 Acceptor Stop Acceptor Einit0 Stop “Parent Region” Stop Stop “Context” Regioning:Segmentation and Counting
Regioning: Simplified • A region includes the sequence to count • A region specifically defines where a model should be counted • The accessor needs no knowledge of strand, regions are reverse complemented on instantiation. • Simply, count from region start to region end on the provided string
Estimation:General Idea • Smoothing • Each model is given a smoother • Normalization • Scoring
Smoother • smoothAref ( ), smoothHref ( ) - smooth the counts for the given parameters
Duration • countFeature ( ) - count the feature duration in the model • smooth ( ) - smooth the counts of the distribution using your smoothers • normalize ( ), score ( ) - convert your counts to scores
Emission • init( ) - Initialize internal variables • clear( ) - Zero out all parameters • countRegion ( ), countNullRegion ( ) - Count a region. • smooth ( ) - Use your smoothers to smooth the data. • normalize ( ), score ( ) - Convert parameters to probabilities or scores. • outputPrepare ( ) - Set the parameter string
Putting it All Together sub _countString { my ($this, $region, $null) = @_; my $buck; if($null) { $buck = $this->nullCounts } else { $buck = $this->posCounts } my $start = $region->start; my $length = $region->end - $region->start + 1; my $weight = $region->weight; my $context = $region->context; my $order = $this->order; my $strRef = $region->strRef; for my $pos (0 .. $length-1) { my $nmer = substr($$strRef, $start+$pos-$order, $order+1); $buck->[$pos+$context]->{$nmer} += $weight; } }
Performance • Runs in about 1-2 hours on the whole genome • Takes up <2GB memory (keeps entire sequence in memory) • Further optimizations can be applied
Prognosis • Running tests now with Randy • Releasing testing version to another lab • Lower-level testing inside the lab • Available on CPAN by the end of the year
Next Predicting skipped exons!