Download

1 / 30
550 likes | 1.65k Views
Atherosclerosis. Jana Novotná Department of Biochemistry 2nd Faculty of Medicine Charles University. Atherosclerosis. Disease of cardiovascular system affecting vessel wall. It leads to the narrowing of arteries or complete bloc k age.

E N D
Atherosclerosis Jana Novotná Department of Biochemistry 2nd Faculty of Medicine Charles University
Atherosclerosis • Disease of cardiovascular system affecting vessel wall. • It leads to the narrowing of arteries or complete blockage. • Its main components are endothelial disfunction, lipid deposition, inflammatory reaction in the vascular wall. • Remodeling of vessel wall.
Atherosclerosis • Intense cross-talk between EC, VSMC, plasma-derived inflammatory cells, lymphocytes (involves array of chemokines, cytokines, growth factors). • Attraction of cells to the sites of atherosclerotic lesion. • Migration, proliferation, apoptosis, excess production of extracellular matrix.
Arterial wall • Normally arterial endothelium repels cells and inhibits blood clotting. • The lumen of healthy arterial wall is lined by confluent layer of endothelial cells.
Arterial wall • Intima (subendothelial layer) • Media (middle layer) with smooth muscle cells (VSMC) • Adventitia (outer layer) with connective tissue and nerves
Arterial wall • Endothelium controls important function: 1. the ability of blood vessels to dilatate (vasodilatation) 2. the ability of blood vessels to constrict (vasoconstriction) • Endothelium regulates tissue and organ blood flow, releases variety substances to control vasomotor tone: • prostacyclines • hyperpolarizing factor • endothelin • NO
Arterial wall • Exercise is an important mechanical stimulus mediated by shear stress to increased blood flow. • Shear stress(t) is a biomechanical lateral force that is determined by blood flow, vessel geometry and fluid viscosity. • The parallel frictional drag force of shear stress is one of the important blood flow-induced mechanical stresses acting on the vessel wall. • Shear stress activates the endothelium and induces nitric oxide release, which causes vasorelaxation. Patel, A., Honoré, E.: Polycystins and renovascular mechanosensory transduction. Nature Reviews Nephrology6:530, 2010
Arterial wall • In the case of intact endothelium, the stimulus for vasodilatation are: • mechanical stimulation by blood flow • catecholamines, bradykinin, platelets-released serotonin stimulate specific receptors • In the case of endothelium disfunction: • direct vasoconstrictor action of the stimuli on the VSMC outweighs the endothelium-dependent vasodilatator effect • this action leads to paradoxial vasoconstriction (Hypercholesterolemia and other cardiovascular risk factors are associated with endothelial disfunction).
Nitric oxide (NO) • Factor which controls vasodilatation: endothelium-derived relaxing factor (EDRF) • The activity of eNOS is controlled by intracellular Ca2+. • eNOS – endothelial constitutive enzyme (continually expressed) and iNOS, inducible, from VSMC. • NO cause vasodilatation via stimulation guanylate cyclase and cGMP production (second messenger for many important cellular functions, particularly for signaling smooth muscle relaxation).
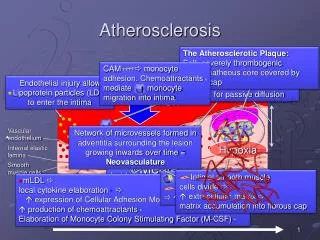
The development of atherosclerosis • The key event – damage to the endothelium caused by excess of lipoproteins, hypertension, diabetes, components of cigarette smoke. • Endothelium becomes more permeable to lipoproteins. • Lipoproteins move below the endothelial layer (to intima). • Endothelium loses its cell-repelent quality. • Inflammatory cells move itno the vascular wall.
Triggers for inflammation in atherosclerosis • LDL retained in the intima (in part by binding to proteoglycan) undergoes oxidative modification. • Lipid hydroperoxides, lysophospholipides, carbonyl compounds localize in the lipid fraction. • Oxygen free-radicals inactivate NO rapidly. • NO + superoxide (O2.-) peroxynitrite (ONOO-). • NO has no longer vasoprotective function.
The development of atherosclerosis • Disfunctional endothelium express adhesion molecules – selectins, mediated the „rolling“ interaction of cells. • The key molecule - vascular cell addhesion molecule-1 (VCAM-1) promotes monocytes adhesion (precursors of macrophages). • Addhering cells are stimulated by monocyte chemoattractant protein-1 (MCP-1). • Monocytes cross the endothelium, settle down in the intima.
NO inactivation due to oxidative stress • Xanthin oxidase and NADH/NADPH oxidase produces of O2.- in macrophages after stimulation with angiotensin II (a key mediator of oxidative stress in vascular wall). • In the early stage of process, ROS are released from endothelium. • In the later atherogenetic process, ROS are produced by macrophages in thickening vessel wall. Schächinger V., Zeiher A.M.: Nephrol Dial Transplant (2002): 2055
Triggers for inflammation in atherosclerosis • Reduced NO bioactivity, increased oxidative stress leads to tyrosine nitration of protein in vessel wall. • Nitrotyrosine and oxidized LDL activate transkriptional factor NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells). • NF-kB increases proliferation of VSMC • Reduced NO bioactivity and enhanced oxidative stress stimulate cytokine production (interleukins, TNFa, MCP-1, interferons) and monocytes attraction.
Inflammation in atherosclerosis (mononuclear cells) Endothelial cells undergo inflammatory activation, produce different leukocyte adhesion molecules (VCAM-1). Monocytes penetrate into tunica intima. Their receptor CCR2 interact with MCP-1 • Resident monocytes acquire characteristics of tissue macrophages. • Macrophages express scavenger receptors for oxidized LDL, internalized lipoprotein particles. Macrophages change to foam cells (lipid droplets are accumulated within the cytoplasm). • Foam cells secrete pro-inflammatory cytokines. • This process amplify local inflammation and ROS. Fom the review article: Libby P.: Inflammation in atherosclerosis. Nature 420, 2002: 868-874
Inflammation in atherosclerosis (T-lymhocytes) T-lymphocytes enter the intima facilitated by VCAM-1 and trio chemokines- IP-10 (inducible protein-1), Mig (monokine induced by interferon-g) and I-TAC (interferon-inducibleT-cells a-chemoattractant). Trio chemokines bind to CXCR3 chemokine receptor expressed by T-cells in the atherogenic lesion. T-cells are activated by antigens (Ox-LDL, heat-shock proteins), produce cytokines. Cytokines influence behavior of other cells present in the atheroma. CD154 (protein expressed on activated T-cells) binding to CD40 ligand expressed on macrophages induces the epression of other cytokines, MMPs, tissue factor. Fom the review article: Libby P.: Inflammation in atherosclerosis. Nature 420, 2002: 868-874
Inflammation in atherosclerosis (mast cells) Mast cells infiltrate to the intima. Chemoattractant eotaxin mediate migration of mast cells and interacts with the chemokine receptor CCR3. Resident mast cells in the intima degranulate, release TNF-a, heparin, and enzymes activating proMMPs. Fom the review article: Libby P.: Inflammation in atherosclerosis. Nature 420, 2002: 868-874
The process of atherogenesis – an overview • Damage to endothelium. • LDL entry to intima. • Expression of adhesion molecules (VCAM-1, MCP-1). • Angiotensin II increases VCAM-1 and MCP-1 expression too. • Monocyte and T lymphocyte attachment, cross the endothelium, lodge in intima. • Monocyte stimulation by M-CSF, transformation into resident macrophages.
The process of atherogenesis – an overview • Production of ROS (macrophages), LDL oxidation. • Decrease NO production increased vasoconstriction. • Expression of macrophage scavenger receptor. • Macrophage change to foam cells. • Stimulation of SMC by cytokines. • SMC proliferation, migration. • Foam cells and lipid pool form the centre of developing atherosclerotic plaques.
The process of atherogenesis • Lipid entry into the arterial wall is a key process in atherogenesis. • Hypercholesterolemia – factor for VCAM-1 and MCP-1 induction. • LDL and VLDL are most atherogenic, enter vascular wall more easily. • LDL – in plasma are protected against oxidation by vit. E, ubiquinon, plasma antioxidants (b-carotene, vit. C). • Out of plasma, LDL phospholipides and fatty acids oxidize.
The process of atherogenesis • Activated macrophages produce enzymes – lipoxygenases, myeloperoxidase, NADPH oxidase. • Oxidized LDL are cytotoxic to endothelial cells, mitogenic for macrophages. • Oxidized LDL apolipoprotein apoB100 bind to the scavenger receptor. • Scavenger receptors are not subjected to regulationby intracellular cholesterol level. • Macrophages take up oxidized LDL, overload with lipids.
The process of atherogenesis • Macrophages changed to foam cells. • Foam cells ruptured (apoptosis). • Lipid release to intima and their accumulation becomes centre of atherosclerotic plaques. • The lipid center and fibrous cap are the main parts of a mature atherosclerotic plaque. • Plaque emerges from the structurally changed vascular wall. • So-called vulnerable plaque ruptures easily. • The thrombus formed at the rupture site.
The process of atherogenesis • Plaque growth – periodically accelerated by a cycle of plaque rupture and thrombosis. • This happens: • Active macrophages and T lymphocytes preferentially reside at the edge of the plaque. • Macrophages secrete enzymes degrade extracellular matrix of the cap (MMP – collagenases, gelatinases, and stromelysin) • T-cells activated by macrophages secrete IFN-g and pro-inflammatory cytokines IL-1, IL-2, and TNF-a.
The process of atherogenesis • IFN-g induces macrophage MMP expression. • IFN-g inhibits VSMC proliferation and collagen synthesis which further weakening the cap. • VSMC undergo apoptosis. • After plaque rupture the area is exposed its interior to the blood. • The interior of the plaque is highly thrombogenic – the small-molecular-weight glycoprotein (tissue factor) initiates the extrinsic clotting cascade. • Tissue factor complexes with factor VII/VIIa, factor IX and X are activated. • Platelets are activated and thrombus forms quickly on the surface of a ruptured plaque. • Thrombus completely occludes the arterial lumen. It cause tissue necrosis (myocardial infarction or brain stroke).
Cardiovascular risk and its assesment • Plasma concentration of lipoproteins (LDL-cholesterol, HDL-cholesterol, and triacylglycerols) – 5.2 mmol/L (200 mg/dL) increases the risk. • Optimal level of LDL-cholesterol - 2.6 mmol/L (100 mg/dL)
Cardiovascular risk and its assesment The risk of atherosclerotic event can be decreased by: • Cholesterol low diet • Exercise • Smoking cessation • Control of high pressure • Drugs statins, fibrates (fibric acid), ezetimibe, antioxidants Statins – inhibit intracellular cholesterol synthase (HMG-CoA reductase). Fibrates - lower plasma cholesterol by stimulating LPL, decreasing TAG concentration, and increasing HDL. Ezetimibe – inhibitor of intestinal cholesterol transporter. b-carotene, a-tocoferol, vitamin C (such as these contained in fruits or their combinations) have preventive benefit, protect LDL against oxidation.
Schächinger V., Zeiher A.M.: Nephrol Dial Transplant (2002): 2055