Download
1 / 16
180 likes | 431 Views
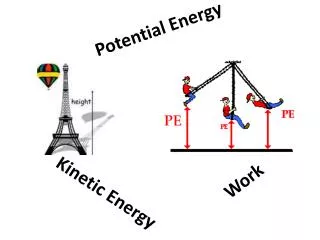
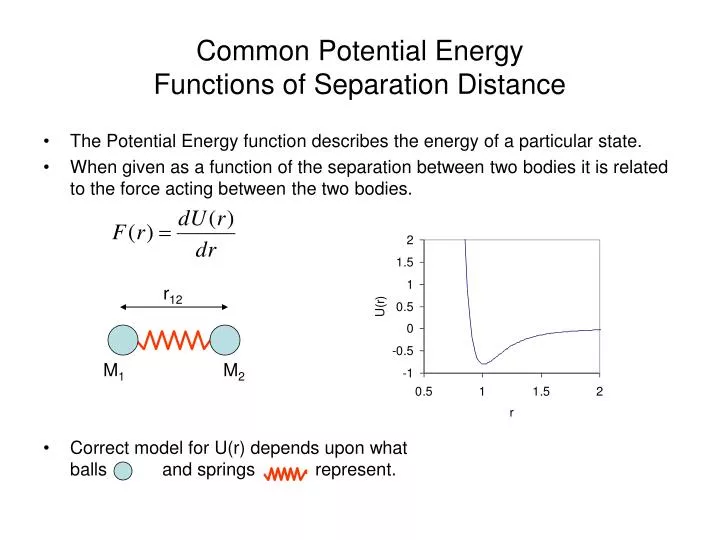
Common Potential Energy Functions of Separation Distance. The Potential Energy function describes the energy of a particular state. When given as a function of the separation between two bodies it is related to the force acting between the two bodies.

E N D
Common Potential Energy Functions of Separation Distance • The Potential Energy function describes the energy of a particular state. • When given as a function of the separation between two bodies it is related to the force acting between the two bodies. • Correct model for U(r) depends upon what balls and springs represent. r12 M1 M2
H H O H O H H O H Common Potential Energy Functions of Separation Distance • For molecular models Balls represent nuclei and Springs represent either bonds or weaker interatomic potentials (coulombic attraction, hydrogen bonds ,Van der Waals attraction etc.)
Common Potential Energy Functions of Separation Distance • In meso-scale PMMA case what are Balls and Springs?
Common Potential Energy Functions of Separation Distance • In meso-scale PMMA case what are Balls and Springs?
Common Potential Energy Functions of Separation Distance • In meso-scale PMMA case what are Balls and Springs?
Common Potential Energy Functions of Separation Distance • In meso-scale PMMA case what are Balls and Springs? do not represent a single covalent bond.
Lennard-Jones • Originally developed to describe force between non-bonded molecules. • Attraction term is the Van der Waals potential of interacting dipoles. • Note that dipoles may only exist temporarily as is the case for nonpolar atoms/molecules such as Helium and Methane. • Repulsive term is supposedly due to repulsion of overlapping orbitals. But this functional form should be exponential (see Morse Potential). • s is effective Van der Waals radius of molecule
Lennard-Jones Potential between non-bonded molecules • Lennard-Jones Parameters for typical non polar molecules. • Values can be found for gaseous molecules in physical chemistry text books. • Used in calculating second virial coefficient perturbation of ideal gas law.
Estimating Lennard-Jones between molecules • s is simply Van der Waals radius of molecules. • Can be estimated using bond additive methods • eg Zhou “Fast calculation of van der Waals volume as a sum of atomic and bond contributions and its application to drug compounds.” Zhao YH, Abraham MH, Zissimos, AM. J Org Chem. 2003 Sep 19;68(19):7368-73 • Or easily calculated using available quantum or molecular modeling software • eg MOPAC, MM2 etc. • e can be estimated using dipole moments of molecules and Gibbs Ensemble formula. • For dislike molecules use Lorentz-Berthelot combination rules.
Using Lennard-Jones Potential to describe bond between two atoms. • s and e can be estimated from Average bond length rbond and bond dissociation energy Ebond. • Typical values for rbond and Ebond can be found in most Organic chemistry texts. Specific calculation requires quantum calculation eg MOPAC etc. • Assume F(r) = 0 at r=rbondand U(r=rbond) – U(r=∞)=Ebond
Shifted-Lennard-Jones • Potential Function • f(r) chosen so both Force and Potential Energy is zero at r=rcutoff • Typically f(r) = a*r+b with rcutoff ~ 2.5*s • Permits clipping of U(r) at short distance without discontinuity in force. • Used for molecular modeling • Calibration is similar to Lennard-Jones Potential
Morse Potential • Leonard-Jones is not good for real bonds. Does not correctly represent both bonding energy Ebond and vibrational strength “k” (eg U(r)=0.5*k*(r-r0)2 ) at bottom of well. • Better “simple” potential is Morse potential. • Parameter “a” controls strength of vibration at bottom of well. • Found from vibrational spectrum frequency of bond.
Proposal of Ways to Estimate U(r) for Mesoscale non-bonded PMMA • Interaction potential between non-bonded groups determined from Lennard-Jones potential between two methy-methacrylate monomers or model monomer units. • Use MOPAC etc. to determine molecular radius and molecular dipole moment. • Confirm values using macroscopic experiments from literature if available. melting point, boiling point, density, Gas/Liquid virial coefficients, yadda yadda ...
Proposal of Ways to Estimate U(r) for Mesoscale bonds in PMMA • Canned Molecular modeling (MM2, Charmm, Amber, Alchemy etc.) and semi-empirical quantum programs (MOPAC etc.) can generate reasonably accurate potential energies for large molecules. • They typically do this for a given conformations • They also can minimize Energy to find lowest energy conformation. • Proposal: • Determine lowest energy conformation for 3-5 MW PMMA. • Stretch chain along some coordinate “r” and calculate new U(r).
Proposal of Ways to Estimate U(r) for Mesoscale bonds in PMMA • Potential Problems: • Coordinate “r” is not a simple coordinate. • It probably does not involve stretching of covalent bonds but rather twisting of bonds. • How to get “r”? • Many different conformations are sampled at each “r “ this introduce an Entropy contribution • Also in statistical mechanics sense U(r) really should be the Free Energy and not potential energy.
Entropic Springs • Systems that have degeneracies along a coordinate transformation possess entropic contributions to Force field. • Classic example are isolated polymer chains in perfect solvent. • Because the solvent is perfect the DU with any conformational change is zero. • However there is a resistive force to stretching the chain away from <R2> • This force is due to the lower entropy (fewer possible conformations) in the streched position than in the coiled position.