Download

1 / 24
240 likes | 441 Views
VIII. NEURODEGENERATIVE DISEASES. - Are disorders characterized by the cellular degeneration of subsets of neurons that typically are related by function, rather than by physical location in the brain.

E N D
- Are disorders characterized by the cellular degeneration of subsets of neurons that typically are related by function, rather than by physical location in the brain. • Many of these disorders are associated with the accumulation of abnormal proteins, which serve as histologic hallmarks of specific disorders • An important but unanswered question is why these abnormal proteins tend to affect particular neurons
, since the involved proteins are widely expressed throughout the nervous system • The possible explanation that subtle differences among subtypes of neurons are presumed to explain why particular neurons are affected in specific disorders. - The clinical manifestations of degenerative diseases are dictated by the pattern of neuronal dysfunction:
A. Those affect the cortical neurons result in dementia; B. Those affect basal ganglia neurons result in movement disorders C. Those that affect the cerebellum result in ataxia Note: Although many degenerative diseases have primary targets, other brain regions are often affected later in the course of the illness
Dementia : = is defined as the development of memory impairment and other cognitive deficits severe enough to decrease the affected person's capacity to function at the previous level despite a normal level of consciousness. - It arises during the course of many neurodegenerative diseases; but it also accompany other diseases that injure the cerebral cortex such as:
1. Infections a. Prion disease & neurosyphilis b. HIV associated neurocognitive disorder c. Chronic meningitis 2. Vascular and traumatic diseases • Multifocal cerebral infarction • b. Chronic traumatic encephalopathy
3. Metabolic and nutritional diseases - Thiamine , Vit B12 and Niacin deficiencies 4. Miscellaneous such as toxicity from lead and manganese
I. Alzheimer Disease (AD) - Is the most common cause of dementia in the elderly population manifests with the insidious onset of impaired higher intellectual function, altered mood and behavior. - Later, this progresses to disorientation, memory loss, and aphasia, findings indicative of severe cortical dysfunction, - And over another 5 to 10 years, the patient becomes profoundly disabled, mute, and immobile.
Death usually occurs from intercurrent pneumonia - Age is an important risk factor for AD; a. The incidence is 3% in persons 65 to 74 years old, b. 9% in 75 to 84 years old to 47% in those older than 84 • Most cases of AD are sporadic • 5% to 10% are familial. • Sporadic cases rarely present before 50 years of age, but early onset is seen with some heritable forms
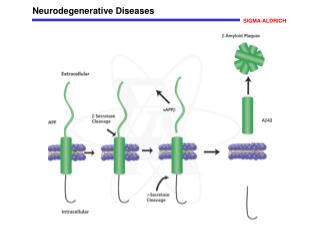
PATHOGENESIS : - Study of the familial forms of AD supports a model in which Aβamyloid, accumulates in the brain over time, initiating a chain of events that result in AD. 1- Aβ is created when the transmembrane protein amyloid precursor protein (APP) is sequentially cleaved by the enzymes β-amyloid converting enzyme (BACE) and γ-secretase - APP also can be cleaved α-secretase and γ-secretase, which liberates a different peptide that is nonpathogenic
.How deposits of Aβcause manifestation 1. While large deposits are a feature of end-stage AD, small aggregates of Aβ may also be pathogenic, as a. They alter neurotransmission and are b. Toxic to neurons and synaptic endings found in the superficial cerebral cortex,
Large deposits, in the form of plaques,: 1. elicite a local inflammatory response Lead to neuronal death 2. And may cause altered region-to-region communication through mechanical effects on axons and dendrites.
3. Aβamyloid also leads to hyperphosphorylation of the neuronal microtubule binding protein tau and this increased level of phosphorylation causes tau to redistribute from axons into dendrites and cell bodies, where it aggregates into tangles, which also contribute to neuronal dysfunction and cell death.
Familial forms 1. Mutations in APP or in components of γ-secretase (presenilin-1 or presenilin-2) lead to familial AD by increasing the rate at which Aβ is generated. 2. The APP gene is located on chromosome 21, - The risk of AD is higher in Down syndrome: an extra copy of the APP gene, is present in patients with trisomy 21
3. The other major genetic risk factor is a variant of apolipoprotein E called Εε4 (ApoE ε 4). • Each ApoE ε 4 allele that is present increases the risk of AD by approximately 4 fold and also appears to lower the age of onset. - How ApoE ε 4 influences Aβ accumulation is unknown; a. It may increase Aβ aggregation or deposition, b. Or decrease Aβ clearance
Macroscopic examination of the brain 1. Variable degree of cortical atrophy, most severe in the frontal, temporal, and parietal lobes. 2. With significant atrophy, there is compensatory ventricular enlargement (hydrocephalus ex vacuo). At the microscopic level, AD is diagnosed by the presence • Of Amyloid plaques (an extracellular lesion) 2. And neurofibrillary tangles (an intracellular lesion)
. Note: - These lesions may also be present to a lesser extent in the brains of elderly non-demented persons, so the current criteria for a diagnosis of AD are based on a combination of clinical and pathologic features. • There is a fairly constant progressive involvement of different parts of the brain.:
Pathologic changes (specifically plaques, tangles, and the associated neuronal loss and glial reaction) a. Are evident first in the entorhinal cortex b. Then in the hippocampal formation, c. And finally in the neocortex.
Neuritic plaques : • Are focal, spherical collections of dilated, tortuous, silver-staining neuritic processes (dystrophic neurites), often around a central amyloid core • Range in size from 20 to 200 μm in diameter - Plaques can be found in the hippocampus and amygdala as well as in the neocortex
- There usually is relative sparing of primary motor and sensory cortices until late in the disease course. 2. Diffuse plaques: - Are Aβ deposits lacking the surrounding neuritic reaction, typically are found in the superficial cerbral cortex, basal ganglia , and the cerebellar cortex
2..Neurofibrillary tangles : - Are bundles of paired helical filaments visible as basophilic in the cytoplasm of the neurons that displace or encircle the nucleus; - Tangles can persist after neurons die, becoming a form of extracellular pathology and are commonly found in:
a. In the entorhinal cortex, b. In pyramidal cells of the hippocampus and amygdala, • The basal forebrain, and the raphe nuclei. - A major component of paired helical filaments is abnormally hyperphosphorylatedtau - Tangles are not specific to AD, being found in other degenerative diseases