Download
1 / 1
10 likes | 181 Views
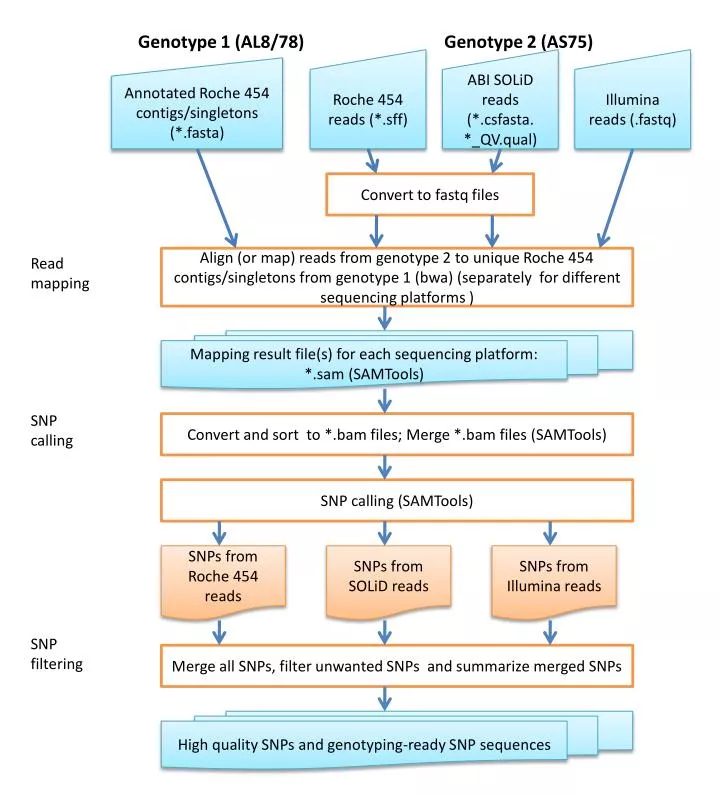
Genotype 1 (AL8/78). Genotype 2 (AS75). Roche 454 reads (*.sff). ABI SOLiD reads (*. csfasta . *_ QV.qual ). Illumina reads (. fastq ). Annotated Roche 454 contigs /singletons (*. fasta ). Convert to fastq files. Read mapping.

E N D
Genotype 1 (AL8/78) Genotype 2 (AS75) Roche 454 reads (*.sff) ABI SOLiD reads (*.csfasta. *_QV.qual) Illumina reads (.fastq) Annotated Roche 454 contigs/singletons (*.fasta) Convert to fastq files Read mapping Align (or map) reads from genotype 2 to unique Roche 454 contigs/singletons from genotype 1 (bwa) (separately for different sequencing platforms ) Mapping result file(s) for each sequencing platform: *.sam (SAMTools) SNP calling Convert and sort to *.bam files; Merge *.bam files (SAMTools) SNP calling (SAMTools) SNPs from Roche 454 reads SNPs from SOLiD reads SNPs from Illumina reads SNP filtering Merge all SNPs, filter unwanted SNPs and summarize merged SNPs High quality SNPs and genotyping-ready SNP sequences