Download

1 / 20
200 likes | 320 Views
SynaMer A New Application for Rapid Identification of Overlapping n-mers From Sequence Reads June 2006. Synamatix team - Introductions. Colin Hercus CTO Poh Yang Ming Bioinformatics Research Team Member Arif Anwar VP. Summary of Agenda. Overview of Genome assembly Key bottlenecks

E N D
SynaMer A New Application for Rapid Identification of Overlapping n-mers From Sequence Reads June 2006
Synamatix team - Introductions • Colin Hercus • CTO • Poh Yang Ming • Bioinformatics Research Team Member • Arif Anwar • VP
Summary of Agenda • Overview of Genome assembly • Key bottlenecks • Introducing SynaMer: • A solution for rapidly finding longer overlapping n-mers • The method • The results • Discussion
Ab initio • genome assembly • Overlap-layout-consensus • Needs high • sequence coverage • No requirement for • closely related genome • Comparative genome • assembly • Alignment-layout-consensus • Requires a closely • related genome • High speed sequence read • to genome mapping is • required • Less dependent • on overlap finding 2 major approaches
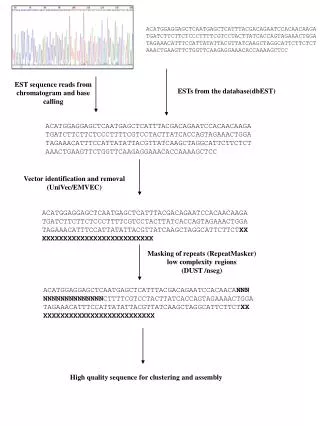
Identified bottlenecks for Ab initio • Typical genome assembly process flow • Sequence reads/Fragments • Vector trimming • Overlapping • Contig/Supercontig/Scaffold generation • Finishing • Final Genome • User* identified major bottleneck in n-mer finding: • Performance • Preference for longer n-mers • IT Hardware requirements User* - Major US Genome Research Institute
Task to accomplish • Original user data set and requirement was: • To find all overlapping exact 100-mers in 50million 1kb sequencing reads – i.e. 50 Billion bp • Report n-mers that have a frequency >2 and <m • Using conventional software and approaches the user took 500hrs and 1.5TB of disc space to find all 100-mer overlaps • Hence standard approach limits usage to 32mers • Longer mers help bridge repetitive and low-complexity regions
Long v Short n-mersadvantages and disadvantages 100 mer +ve Fewer false positives Improvement in final assembly Errors in reads may lead to false negatives Slow to process with conventional software -ve
Explanation of advantages A A shorter overlap results in more false positives Low-complexity region B A longer overlap results in less false positives Final assembly improved
Synamer: A solution for rapid identification of longer n-mers • Synamer finds overlapping sequences given a defined “n” with a range of frequency of occurrence in the sequence set • It is similar to a class of tools in genome assembly called “overlappers” • 2 well known overlappers are: • UMD Overlapper • Roberts M et. al.(2004) Bioinformatics 20(18):3363-3369 • KI Overlapper • Tammi MT et.al., (2003) NAR 31(15):4663-4672
How Synamer works • Given a mer length of “n” • Extract a n-mer at each position within a read • Compare the n-mer and reverse complement, to report palindromes • Index n-mers and their location within reads • For each n-mer within a user defined frequency range report the n-mer and locations
SynaMer data • Input: • Text file of the reads • Parameters: • n (default 96, maximum of 128) • Frequency range (default of 2 to 50) • Memory usage (Default to available memory) • Temporary file location • Output Format: • Text or binary • n-mer Frequency Palindrome direction read ID:location
Test cases • User case 1: 30million 1kb reads finding exact 96mer took approximately 5hrs to process, with less than 200GB temporary disk space on a dual CPU Itanium • Compared to 500hrs and over 1.5TB of disk space • Use case 2: • Brucella_suis 1330, 36080 900bp reads (http://www.tigr.org/tdb/benchmark/) • Tests were conducted with a range of n-mer with frequency of minimum of 2 to 120. • n-mer range of: 12, 24, 36, 48, 60, 72, 84, 96, 108, 120 • Average execution time measured with 6 replicates
Brucella suis - results • Majority of the patterns are at frequency of 2-50 • More pattern at higher n-mer • Longer n-mer would be more specific and less false positive
Distribution of overlapping sequence with frequency Higher level of repeats in more complex genomes leads to increased benefits from using longer n-mers
Sample Output • At 96-mer: • TTTCATAAAGCCGCTTTGCACCATAAAGCGCGTCGCCGGTGCTGCCTGTGGTGCCGTAGAAAGTCCAGCCTTCCTCCGCCATCAGGAAATCAACCACTGAAACGGAAA 5 33984:395 25036:255 17186:435 -5741:85 5184:181 • TTTCATAAACCTGACCCTGATTCGCCGCACCATCGCCGAAATAGGTCAGCGAAACGGATTTATTCTCACGATAGTGATTGGCGAAGGCCAACCCCGTACCGAGCGAAA 8 30929:163 28279:329 25051:228 -22556:257 -14554:249 -12303:286 15820:325 6770:434 • TTTCATAAAACCTAAATAATATAGAATATATTTTTTAATTTACTCCCACAAAAATTGATATTTATAAAATAAAAAATCCCAATCTGTAAATCCCAATAATTTTACAAA 4 32618:184 -9587:456 9891:617 8902:369 • TTTCAGTTTCTCAAGCAAACCCTTTATGACATTGCATCTTTGCTGGTGTTTTTCGCCAATGTTGCATTTTGTTTCTCAATTGTAGCGCAAGCAAATGCGGCTTGAAAA 5 26073:487 -21045:262 22952:244 12603:19 6640:383 • The numbers before the “:” are the ordinal position of the reads in the file
Sample contig • To show validity of the result • GBUAS15TR and GBUCA37TF • Detected overlap at 96-mer – shown below: • At position 188 on GBUAS15TR and 811 on GBUCA37TF • They can be joined to a 1.5kbp contig, with consensus
Conclusions • For 30million 1kb reads took 5 hours on a dual CPU itanium machine, with temporary file size less than 200GB • Time consumed to find overlapping sequences for 33000 900bp reads of a bacterial WGSS reads took less than 20s • 100 fold faster than conventional method • Allows use of longer n-mers • Potentially increases quality of assembly • SynaMer will be made released as a product later this Summer
Questions and Follow up • Please send questions to: tech@synamatix.com • Webcast will be available online in 24hrs at • www.mgrc.com.my • Paper accompanying this webcast will be sent to all attendees • If you are interested in testing SynaMer when it is released please email: enquries@synamatix.com
Thank you! tech@synamatix.com