Download

1 / 23
330 likes | 897 Views
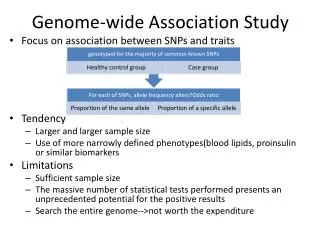
Genome-Wide Association Study (GWAS). Presented by Karen Xu. What you need to know. Basic genetic concepts behind GWAS Genotyping technologies and common study designs Statistical concepts for GWAS analysis Replication, interpretation and follow-up of association results.

E N D
Genome-Wide Association Study (GWAS) Presented by Karen Xu
What you need to know • Basic genetic concepts behind GWAS • Genotyping technologies and common study designs • Statistical concepts for GWAS analysis • Replication, interpretation and follow-up of association results
Central Goal of Human Genetics • To identify genetic risk factors for common, complex diseases
Goal of GWAS • To use genetic risk factors to predict who is at risk • Identify the biological underpinnings of disease susceptibility for developing new prevention and treatment strategies
Application in pharmacology • Identifying DNA sequence variations associated w/ drug metabolism and efficacy as well as adverse effects • Example, warfarin---determining the appropriate dose • Personalized medicine
Concepts underlying the study design • SNP---single nucleotide polymorphism • Single base pair changes in the DNA sequence that occur with high frequency in the human genome • SNP (common) vs. Mutation (rare) • Cystic fibrosis---mutations in the CFTR gene • Linage analysis---genotyping families affected by cystic fibrosis using a collection of genetic markers across the genome and examining how these genetic markers segregate w/ the disease across multiple familes
Common Disease Common Variant Hypothesis • Common disorders are likely influenced by genetic variation that is also common in the population • 1. If common genetic variants influence disease, the effect size (or penetrance) for any one variant must be small relative to that found for rare disorders. • 2. If common alleles have small genetic effects (low penetrance), but common disorders show heritability (inheritance in families), then multiple common alleles must influence disease susceptibility.
Figure 1. Spectrum of Disease Allele Effects. Bush WS, Moore JH (2012) Chapter 11: Genome-Wide Association Studies. PLoSComputBiol 8(12): e1002822. doi:10.1371/journal.pcbi.1002822 http://www.ploscompbiol.org/article/info:doi/10.1371/journal.pcbi.1002822
Capturing Common Variation • 1. location and density of commonly occurring SNPs is needed to identify the genomic regions and individual sites that must be examined by genetic studies • 2. population-specific differences in genetic variation must be cataloged so that studies of phenotypes in different populations can be conducted with the proper design • 3. correlations among common genetic variants must be determined so that genetic studies do not collect redundant information
International HapMap Project • Used a variety of sequencing techniques to discover and catalog SNPs in European descent populations, the Yoruba populations of African origin, Han Chinese individuals from Beijing, and Japanese individuals from Tokyo • Has since been expanded to include 11 human populations
Linkage Disequilibrium • A property of SNPs on a contiguous stretch of genomic sequence that describes the degree to which an allele of a SNP is inherited or correlated with an allele of another SNP within a population • Linkage between markers on a population scale
Figure 2. Linkage and Linkage Disequilibrium. Bush WS, Moore JH (2012) Chapter 11: Genome-Wide Association Studies. PLoSComputBiol 8(12): e1002822. doi:10.1371/journal.pcbi.1002822 http://www.ploscompbiol.org/article/info:doi/10.1371/journal.pcbi.1002822
Direct vs. Indirect Association • LD creates two possible positive outcomes from a genetic association study • 1. direct association----the SNP influencing a biological system that leads to the phenotype is directly genotyped in the study • 2. Indirect association----the influential SNP is not directly typed, but instead a tag SNP in high LD with the influential SNP is typed • Therefore, a significant SNP association from a GWAS should not be assumed as the causal variant
Genotyping Technologies • Chip-based microarray technology • Illumina, NA molecules and primers are first attached on a slide and amplified with polymerase so that local clonal DNA colonies, later coined "DNA clusters", are formed. To determine the sequence, four types of reversible terminator bases (RT-bases) are added and non-incorporated nucleotides are washed away. A camera takes images of the fluorescently labeled nucleotides, then the dye, along with the terminal 3' blocker, is chemically removed from the DNA, allowing for the next cycle to begin.
Study Design • Case control vs. quantitative design • Two primary classes of phenotypes: categorical or quantitative • From the statistical perspective, quantitative traits are preferred, but not required for a successful study
Association Test • 1. single-locus analysis • When a well-defined phenotype has been selected for a study population, and genotypes are collected using sound techniques, the statistical analysis can begin • Quantitative traits----ANOVA (analysis of variance)---null hypothesis is that there is no difference between the trait means of any genotype group • Dichotomous case/ control traits are analyzed using logistic regression---null hypothesis---there is no association between the phenotype and genotype • http://luna.cas.usf.edu/~mbrannic/files/regression/Logistic.html
Statistical replication • Replication studies should be conducted in an independent dataset drawn from the same population as GWAS • Once an effect is confirmed in the target population, other populations may be sampled to determine if the SNP has an ethnic-specific effect • Identical phenotype criteria should be used in both GWAS and replication studies • A similar effect should be seen in the replication set from the same SNP, or a SNP in high LD with the GWAS-identified SNP
Meta-analysis of multiple analysis results • Meta-analysis developed to examine and refine significance and effect size estimates from multiple studies examining the same hypothesis in the published literature • However, it is rare to find multiple studies that match perfectly on all criteria • Study heterogeneity is often statistically quantified in a meta-analysis to determine the degree to which studies differ.
Data Imputation • To conduct a meta-analysis properly, the effect of the same allele across multiple distinct studies must be assessed. This can prove difficult if different studies use different genotyping platforms (which use different SNP marker sets). As this is often the case, GWAS datasets can be imputed to generate results for a common set of SNPs across all studies. Genotype imputation exploits known LD patterns and haplotype frequencies from the HapMap or 1000 Genomes project to estimate genotypes for SNPs not directly genotyped in the study [50].
Logistic regression • Predicting the likelihood that Y is equal to 1 (rather than 0) given certain values of X • Example: we try to predict whether or not small business will succeed based on the number of years of experience the owner has in the field prior to starting the business. We presume that those people who have more experience will be more likely to succeed • As X (the number of years of experience) increases, the probability that Y will be equal to 1 (success in the business) will tend to increase