Download

1 / 53
530 likes | 565 Views
Learn about why tandem mass spectrometry is crucial for identifying proteins. Explore topics such as enzymatic digestion, peptide fragmentation, and unannotated splice isoforms in different organisms. Use advanced search engines for precise peptide identification and functional annotation.

E N D
Generalized Protein Parsimony and Spectral Counting for Functional Enrichment Analysis Nathan Edwards Department of Biochemistry and Molecular & Cellular Biology Georgetown University Medical Center
Why Tandem Mass Spectrometry? • LC-MS/MS spectra provide evidence for the amino-acid sequence and abundance of functional proteins. • Key concepts: • Spectrum acquisition is unbiased by knowledge • Direct observation of amino-acid sequence • Sensitive to small sequence variations • Spectrum acquisition is biased by abundance
Enzymatic Digest and Fractionation Sample Preparation for MS/MS
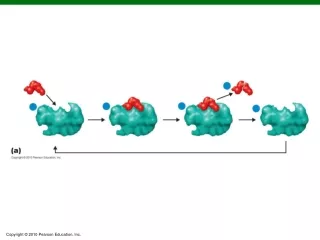
Tandem Mass Spectrometry(MS/MS) Precursor selection
Tandem Mass Spectrometry(MS/MS) Precursor selection + collision induced dissociation (CID) MS/MS
Peptide: S-G-F-L-E-E-D-E-L-K MW ion ion MW 88 b1 S GFLEEDELK y9 1080 145 b2 SG FLEEDELK y8 1022 292 b3 SGF LEEDELK y7 875 405 b4 SGFL EEDELK y6 762 534 b5 SGFLE EDELK y5 633 663 b6 SGFLEE DELK y4 504 778 b7 SGFLEED ELK y3 389 907 b8 SGFLEEDE LK y2 260 1020 b9 SGFLEEDEL K y1 147 Peptide Fragmentation
Unannotated Splice Isoform • Human Jurkat leukemia cell-line • Lipid-raft extraction protocol, targeting T cells • von Haller, et al. MCP 2003. • LIME1 gene: • LCK interacting transmembrane adaptor 1 • LCK gene: • Leukocyte-specific protein tyrosine kinase • Proto-oncogene • Chromosomal aberration involving LCK in leukemias. • Multiple significant peptide identifications
Translation start-site correction • Halobacterium sp. NRC-1 • Extreme halophilic Archaeon, insoluble membrane and soluble cytoplasmic proteins • Goo, et al. MCP 2003. • GdhA1 gene: • Glutamate dehydrogenase A1 • Multiple significant peptide identifications • Observed start is consistent with Glimmer 3.0 prediction(s)
Halobacterium sp. NRC-1ORF: GdhA1 • K-score E-value vs PepArML @ 10% FDR • Many peptides inconsistent with annotated translation start site of NP_279651
Lost peptide identifications • Missing from the sequence database • Search engine strengths, weaknesses, quirks • Poor score or statistical significance • Thorough search takes too long
Peptide Sequence Databases • All amino-acid 30-mers, no redundancy • From ESTs, Proteins, mRNAs • 30-40 fold size and search time reduction • Formatted as a FASTA sequence database • One entry per gene/cluster.
SEQUEST Mascot 28% 14% 14% 38% 1% 3% 2% X! Tandem Combine search engine results • No single score is comprehensive • Search engines disagree • Many spectra lack confident peptide assignment Searle et al. JPR 7(1), 2008
Combining search engine results – harder than it looks! • Consensus boosts confidence, but... • How to assess statistical significance? • Gain specificity, but lose sensitivity! • Incorrect identifications are correlated too! • How to handle weak identifications? • Consensus vs disagreement vs abstention • Threshold at some significance? • We apply "unsupervised" machine-learning.... • Lots of related work unified in a single framework.
PepArML Workflow • Select high-quality IDs • Guess true proteins from search results • Label spectra & train • Calibrate confidence • Guess true proteins from ML results • Iterate! • Estimate FDR using (external) decoy
PepArML Meta-Search Engine X!Tandem, KScore, OMSSA, MyriMatch, Mascot (1 core). NSF TeraGrid 1000+ CPUs Heterogeneous compute resources X!Tandem, KScore, OMSSA, MyriMatch. Secure communication Edwards Lab Scheduler & 80+ CPUs Amazon AWS Scales easily to 250+ simultaneoussearches Single, simplesearch request
PeptideMapper Web Service I’m Feeling Lucky
PeptideMapper Web Service I’m Feeling Lucky
PeptideMapper Web Service • Suffix-tree index on peptide sequence database • Fast peptide to gene/cluster mapping • “Compression” makes this feasible • Peptide alignment with cluster evidence • Amino-acid or nucleotide; exact & near-exact • Genomic-loci mapping via • UCSC “known-gene” transcripts, and • Predetermined, embedded genomic coordinates
Systems Biology molecular biology ↕phenotype molecular biology↕biology Structured High-ThroughputExperiments KnowledgeDatabases • Proteomics • Sequencing • Microarrays • Metabolomics • Localization • Function • Process • Interactions • Pathway • Mutation
Systems Biology molecular biology ↕phenotype molecular biology↕biology Structured High-ThroughputExperiments KnowledgeDatabases FunctionalAnnotation Enrichment • Proteomics • Sequencing • Microarrays • Metabolomics • Localization • Function • Process • Interactions • Pathway • Mutation MathematicalModels
Systems Biology molecular biology ↕phenotype molecular biology↕biology Structured High-ThroughputExperiments KnowledgeDatabases FunctionalAnnotation Enrichment • Proteomics • Sequencing • Microarrays • Metabolomics • Localization • Function • Process • Interactions • Pathway • Mutation MathematicalModels
Why not in proteomics? • Double counting and false positives… • …due to traditional protein inference • Proteomics cannot see all proteins… • …proteins are not equally likely to be drawn • Good relative abundance is hard… • …extra chemistries, workflows, and software • …missing values are particularly problematic
In proteomics… • Double counting and false positives… • Use generalized protein parsimony • Proteomics cannot see all proteins… • Use identified proteins as background • Good relative abundance is hard… • Model differential spectral counts directly
Traditional Protein Parsimony • Select the smallest set of proteins that explain all identified peptides. • Sensible principle, implies • Eliminate equivalent/subset proteins • Equivalent proteins are problematic: • Which one to choose? • Unique-protein peptides force the inclusion of proteins into solution • True for most tools, even probability based ones • Bad consequences for FDR filtered ids
Peptide-Spectrum Matches • Sigma49 – 32,691 LTQ MS/MS spectra of 49 human protein standards; IPI Human • Yeast – 162,420 LTQ MS/MS spectra from a yeast cell lysate; SGD. • X!TandemE-value (no refinement), 1% FDR Spectra used in: Zhang, B.; Chambers, M. C.; Tabb, D. L. 2007.
Many proteins are easy • Eliminate equivalent / dominated proteins • Sigma49: 277 → 60 proteins • Yeast: 1226 → 1085 proteins • Many components have a single protein: • Sigma49: 52 ( 3 multi-protein) • Yeast: 994 (43 multi-protein) • Single peptides force protein inclusion • Sigma49: 16 single-peptide proteins • Yeast: 476 single-peptide proteins
Must eliminate redundancy • Contained proteins should not be selected 37 distinct peptides
Must eliminate redundancy 1.0 1.0 0.8 0.7 0.0 1.0 • Contained proteins should not be selected • Even if they have some probability mass • Number of sibling peptides matter less if they are shared. Single AA Difference
Must ignore some PSMs 1.0 0.0 0.0 0.0 0.0 1.0 • A single additionalpeptideshould not force protein into solution Single AA Difference
Example from Yeast • "Inosine monophosphate dehydrogenase" • 4 gene family • Contained proteins should not be selected • Single peptide evidence for YML056C 1.0 0.6 0.0 1.0
Must ignore some PSMs • Improving peptide identification sensitivitymakes things worse! • False PSMs don't cluster PSMs PSMs 2x Proteins 10%
Must ignore some PSMs • Improving peptide identification sensitivitymakes things worse! • False PSMs don't cluster PSMs PSMs Select Proteins to Explain True PSM% 90% 90%
Must ignore some PSMs • How do we choose? • Maximize # peptides? • Minimize FDR (naïve model)? • Maximize # PSMs?
Generalized Protein Parsimony • Weight peptides by number of PSMs • Constrainunique peptides per protein • Maximize explained peptides (PSMs) • Match PSM filtering FDR to % uncovered PSMs • Readily solved by branch-and-bound • Permits complex protein/peptide constraints • Reduces to traditional protein parsimony
Plasma membrane enrichment • Pellicle enrichment of plasma membrane • Choksawangkarn et al. JPR 2013 (Fenselau Lab) • Six replicate LC-MS/MS analyses each • Cell-lysate (44,861 MS/MS) • Fe3O4-Al2O3 pellicle (21,871 MS/MS) • 625 3-unique proteins to match 10% FDR: • Lysate: 18,976 PSMs; Pellicle: 13,723 PSMs • 89 proteins with significantly (< 10-5) increased counts
Semi-quantitative LC-MS/MS Precursor selection + collision induced dissociation (CID) MS/MS
Semi-quantitative LC-MS/MS Chen and Yates. Molecular Oncology, 2007
Plasma membrane enrichment • Na/K+ ATPase subunit alpha-1 (P05023): • Lysate: 1; Pellicle: 90; p-value: 5.2 x 10-33 • Transferrin receptor protein 1 (P02786): • Lysate: 17; Pellicle: 63; p-value: 2.0 x 10-11 • DAVID Bioinformatics analysis (89/625): • Plasma membrane (GO:0005886) : 29 (5.2 x 10-5) • Transmembrane (SwissProtKW): 24 (1.3 x 10-6) • Transmembrane (SwissProtKW): • Lysate: 524; Pellicle: 1335; p-value: 2.6 x 10-158
HER2/Neu Mouse Model of Breast Cancer • Paulovich, et al. JPR, 2007 • Study of normal and tumor mammary tissue by LC-MS/MS • 1.4 million MS/MS spectra • Peptide-spectrum assignments • Normal samples (Nn): 161,286 (49.7%) • Tumor samples (Nt): 163,068 (50.3%) • 4270 proteins identified in total • 2-unique generalized protein parsimony
Nascent polypeptide-associated complex subunit alpha 7.3 x 10-8