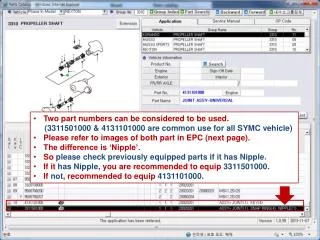
Download
1 / 1
10 likes | 152 Views
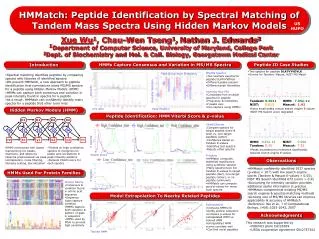
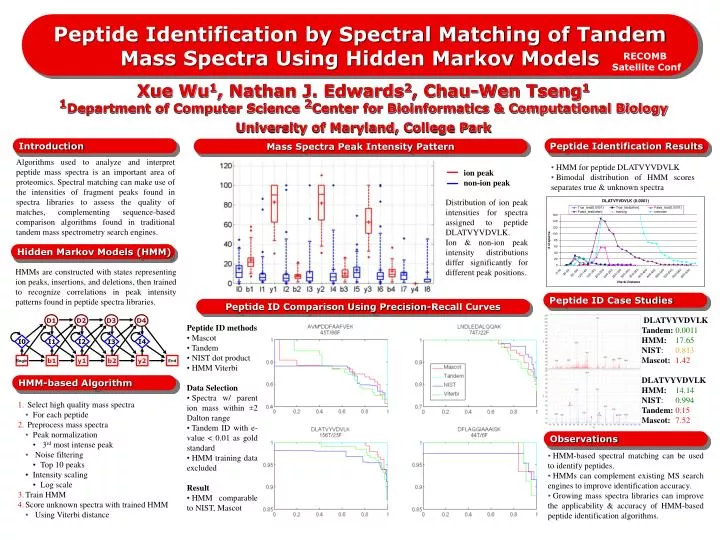
ion peak non-ion peak. D1. D2. D3. D4. I0. I1. I2. I4. I3. Begin. b1. y1. b2. y2. End. Peptide Identification by Spectral Matching of Tandem Mass Spectra Using Hidden Markov Models. RECOMB Satellite Conf.

E N D
ion peak non-ion peak D1 D2 D3 D4 I0 I1 I2 I4 I3 Begin b1 y1 b2 y2 End Peptide Identification by Spectral Matching of Tandem Mass Spectra Using Hidden Markov Models RECOMB Satellite Conf Xue Wu1, Nathan J. Edwards2, Chau-Wen Tseng11Department of Computer Science 2Center for Bioinformatics & Computational Biology University of Maryland, College Park Introduction Peptide Identification Results Mass Spectra Peak Intensity Pattern Algorithms used to analyze and interpret peptide mass spectra is an important area of proteomics. Spectral matching can make use of the intensities of fragment peaks found in spectra libraries to assess the quality of matches, complementing sequence-based comparison algorithms found in traditional tandem mass spectrometry search engines. • HMM for peptide DLATVYVDVLK • Bimodal distribution of HMM scores separates true & unknown spectra Distribution of ion peak intensities for spectra assigned to peptide DLATVYVDVLK. Ion & non-ion peak intensity distributions differ significantly for different peak positions. Hidden Markov Models (HMM) HMMs are constructed with states representing ion peaks, insertions, and deletions, then trained to recognize correlations in peak intensity patterns found in peptide spectra libraries. Peptide ID Case Studies Peptide ID Comparison Using Precision-Recall Curves • DLATVYVDVLKTandem: 0.0011 • HMM:17.65 • NIST: 0.813 • Mascot:1.42 • DLATVYVDVLK • HMM:14.14 • NIST: 0.994 • Tandem: 0.15 • Mascot:7.52 • Peptide ID methods • Mascot • Tandem • NIST dot product • HMM Viterbi • Data Selection • Spectra w/ parent ion mass within ±2 Dalton range • Tandem ID with e-value < 0.01 as gold standard • HMM training data excluded • Result • HMM comparable to NIST, Mascot HMM-based Algorithm • Select high quality mass spectra • For each peptide • Preprocess mass spectra • Peak normalization • 3rd most intense peak • Noise filtering • Top 10 peaks • Intensity scaling • Log scale • Train HMM • Score unknown spectra with trained HMM • Using Viterbi distance Observations • HMM-based spectral matching can be used to identify peptides. • HMMs can complement existing MS search engines to improve identification accuracy. • Growing mass spectra libraries can improve the applicability & accuracy of HMM-based peptide identification algorithms.