Download

1 / 17
170 likes | 372 Views
Cystic Fibrosis. Gary Hoffman Wisconsin Newborn Screening Laboratory. Cystic Fibrosis Review. Most common lethal genetic autosomal recessive disorder Caucasians 1:3,000 African Americans 1:20,000 Hispanics 1:7,500 Asians 1:30,000 Multi-Organ dysfunction

E N D
Cystic Fibrosis Gary Hoffman Wisconsin Newborn Screening Laboratory
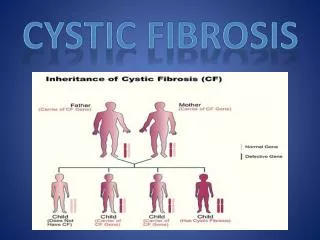
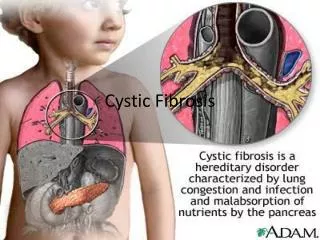
Cystic Fibrosis Review • Most common lethal genetic autosomalrecessive disorder Caucasians 1:3,000 African Americans 1:20,000 Hispanics 1:7,500Asians 1:30,000 • Multi-Organ dysfunction digestive system reproductive system (males) respiratory tract • Clinical manifestations Irreversible lung damage (infections) Severe Malnutrition
Cystic Fibrosis Review (Cont) • Life expectancy 50% die before age 32 • Significant morbidity at any age • Gene Transmembrane Conductance Regulator (CFTR) On long arm of chromosome 7 • lack of salt and water transfer across cell membranes 1,500+ mutations in the CFTR • Confirmatory diagnosis Positive sweat chloride measurements
Wisconsin Experience Randomized Control Trial (RCT) 1985 – 1990 Immunoreactive trypsinogen IRT cutoff – 99.8%tile False negative rate to high 1991 - 1994 Incorporated 2nd tier DNA F508del mutation Polyacrylamide gel with silver staining IRT cutoff – 98.5%tile Significantly reduced false negatives
Wisconsin Experience (cont) Routine screening July 1994 Two tiered algorithm 1st tier – Immunoreactive trypsinogen 2nd tier – Mutation analysis for F508del Initial IRT cutoff for mutation testing – 94th%tile Changes Increased percentile to 96th Changed methodology Roche linear array 23 mutations Hologic (TWT) invader
Mutation Analysis Issues • Problem Specimens • No product formation • Insufficient DNA • Excessive hemolysis - inhibits PCR • Transfusions • Delay in reporting results • 5 to 8 days depending upon PCR testing schedule
Mutation Methods Targeted mutation analysis (most common) Allele specific primers for wild type and mutant bind and amplify small regions around mutation site mutations can be substitutions, insertions, deletions
Mutation Methods FDA approved kits: Luminex (xTAG) Multiple Allele-Specific Primer Extension (ASPE) beads that generate signals when separated by laser flow cytometry. 39, 60, and 71 mutation panel Program for only ACMG 23 panel Open architecture Hologic – InPlex Flurorescence Resonance Energy Transfer (FRET) signal is generated with cleavase ACMG 23 Panel limited hands on time cartridge based
Selection of CF mutations Over 1500 unknown mutations Technology can identify about 100 mutations Population demographics Match panel with genotypes 23 – 40 mutations will identify >95% of the cases Minimum Core – ACMG recommended 23 Disease causing mutations (R117H?) Panels do not need to cover 100% of the disease causing alleles
Mutation Testing Summary Increase specificity and detection sensitivity Minimize false negatives Expedite management and treatment Identification of carriers
Maple Syrup Urine Disease Gary Hoffman Wisconsin Newborn Screening Laboratory
Overview of MSUD Aminoacidopathy of leucine metabolism Primary screening analytes – Leucine/Isoleucine & Valine Primary methodology – Tandem Mass Spectrometry Prevalence Overall population – 1:250,000 Mennonite – 1:400 Clinical symptoms seizures, coma, vomiting, sweet smelling urine, death, developmental delay Treatment: low protein diet Recommended treatment age: 72 hours from birth
Selection of Mutations Gene Branched chain keto acid deydrogenase E1, alpha polypeptide (BCKDHA) Long arm of chromosome 19 40 unknown mutations Disrupt BCKD enzyme from breakdown leucine, valine, and isoleucine Mennonite populations Y438N (Tyr438Asn)
Wisconsin Method Tetra-primer ARMS-PCR Two flanking primers; 2 internal primers Two annealing temps Concentrates the DNA fragments Amplicons separated on agarose gel Homozygote – one band Heterozygote – two bands
Molecular Testing Advantages Support of the screening results Provide for earlier intervention High risk births – immediate testing Inexpensive testing
MSUD SUCCESS STORY Couple known carrier of Y438N mutation Specimen collected at one hour of age Transported to NBS lab within 2 hours Assay setup immediately Result Y438N homozygote (8 hours of age) Dietary treatment at 12 hours of age Outcome One hospitalization in the first week of life Family compliant with therapy At 4 years - no mental or physical issues