Download
1 / 1
10 likes | 121 Views
A Phospho -Peptide Spectrum Library for Improved Targeted Assays Barbara Frewen 1 , Scott Peterman 1 , John Sinclair 2 , Claus Jorgensen 2 , Amol Prakash 1 , Mary Lopez 1

E N D
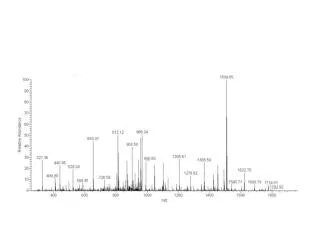
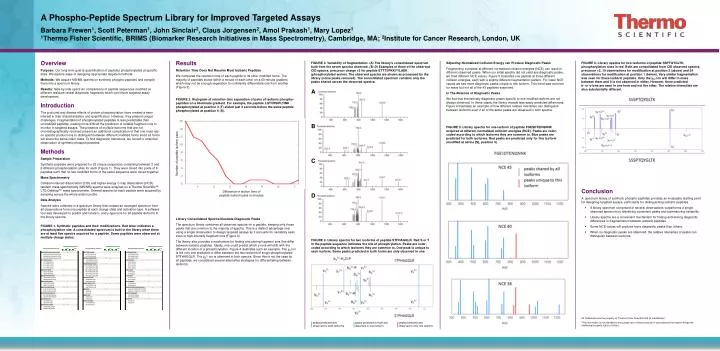
A Phospho-Peptide Spectrum Library for Improved Targeted Assays Barbara Frewen1, Scott Peterman1, John Sinclair2, Claus Jorgensen2, Amol Prakash1, Mary Lopez1 1Thermo Fisher Scientific, BRIMS (Biomarker Research Initiatives in Mass Spectrometry), Cambridge, MA; 2Institute for Cancer Research, London, UK Overview Purpose: Our long-term goal is quantification of peptides phosphorylated at specific sites. We explore ways of designing appropriate targeted methods. Methods: We acquire MS/MS spectra on synthetic phospho-peptides and compile them into a spectrum library. Results: Side-by-side spectrum comparisons of peptide sequences modified at different residues reveal diagnostic fragments which can inform targeted assay development. Introduction The profound and diverse effects of protein phosphorylation have created a keen interest in their characterization and quantification. However, they present unique challenges. Fragmentation of phosphorylated peptides is less predictable than unmodified peptides, making more difficult the prediction of reliable fragment ions to monitor in targeted assays. The presence of multiple isoforms that are not chromatographically resolved present an additional complication in that one must rely on specific product ions to distinguish between different modified forms since all forms will share the same intact mass. To find diagnostic transitions, we turned to empirical observation of synthetic phospho-peptides. Methods Sample Preparation Synthetic peptides were prepared for 25 unique sequences containing between 2 and 6 different phosphorylation sites for each (Figure 1). They were mixed into pools of 8 peptides such that no two modified forms of the same sequence were mixed together. Mass Spectrometry Collision-induced dissociation (CID) and higher-energy C-trap dissociation (HCD) tandem mass spectrometry (MS/MS) spectra were acquired on a Thermo Scientific™ LTQ Orbitrap™ mass spectrometer. Several spectra for each peptide were acquired by sampling across the whole elution profile. Data Analysis Spectra were collected in a spectrum library that creates an averaged spectrum from all observations from one peptide at each charge state and activation type. A software tool was developed to predict and locate b- and y-type ions for all peptide isoforms in the library spectra. Results Retention Time Does Not Resolve Most Isobaric Peptides We compared the retention time of each peptide to its other modified forms. The majority of peptides eluted within a minute of each other on a 60-minute gradient, which may not be enough separation to confidently differentiate one from another(Figure 2). FIGURE 3. Variability of fragmentation. (A) The library’s consolidated spectrum built from the seven spectra observed. (B–D) Examples of three of the observed CID spectra, precursor charge +2 for peptide ETTTSPKKYYLAEK (phosphorylated serine). The observed spectra are shown as processed by the library (noise peaks removed). The consolidated spectrum contains only the peaks shared across the observed spectra. FIGURE 6. Library spectra for two isoforms of peptide SSPTQYGLTK, phosphorylation sites in red. Both are consolidated from CID observed spectra, precursor +2, 19 observations for modification at position 2 (above) and 24 observations for modification at position 1 (below). Very similar fragmentation was seen for these isobaric peptides. Only the y10 ion will differ in mass between them and it is not observed in either. However, three predicted b- or y-ions are seen in one form and not the other. The relative intensities are also substantially different. Adjusting Normalized Collision Energy can Produce Diagnostic Peaks Fragmenting a peptide at different normalized collision energies (NCE) can result in different observed peaks. When our initial spectra did not yield any diagnostic peaks, we tried different NCE values. Figure 5 illustrates one peptide at three different collision energies, each with a slightly different fragmentation pattern. For lower NCE values we see more diagnostic peaks unique to this isoform. This trend was common for many but not all of the 43 peptides examined. In The Absence of Diagnostic Peaks We find that theoretically diagnostic peaks specific to one modified isoform are not always observed. In those cases, the library reveals less-easily predicted differences. Figure 6 illustrates an example of how different relative intensities can distinguish between isoforms even if all of the same peaks are observed in both spectra. A FIGURE 2. Histogram of retention time separation of pairs of isobaric phospho-peptides on a 60-minute gradient. For example, the peptide LQTVHSIPLTINK phosphorylated at position 3 (T) eluted just 3 seconds before the same peptide phosphorylated at position 6 (S). B FIGURE 5. Library spectra for one isoform of peptide FGESDTENQNNK acquired at different normalized collision energies (NCE). Peaks are color-coded according to which isoforms they are common to. Blue peaks are predicted for both isoforms. Red peaks are predicted only for this isoform (modified at serine [S], position 4). C Number of peptide isoform pairs • Conclusion • A spectrum library of synthetic phospho-peptides provides an invaluable starting point for designing targeted assays, particularly for distinguishing isobaric peptides. • A library spectrum comprised of several observations outperforms a single observed spectrum by identifying consistent peaks and summarizing variability. • Library spectra are a convenient mechanism for finding and storing diagnostic differences in fragmentation between isobaric peptides. • Some NCE values will produce more diagnostic peaks than others. • When no diagnostic peaks are observed, the relative intensities of peaks can distinguish between isoforms. D Difference in elution time of peptide isoform pairs in minutes Library Consolidated Spectra Illustrate Diagnostic Peaks The spectrum library combines all observed spectra for a peptide, keeping only those peaks that are common to the majority of spectra. This is a distinct advantage over using a single observation to design targeted assays as it accounts for variability seen even in high-intensity fragment ions (Figure 3). The library also provides a mechanism for finding and storing fragment ions that differ between isobaric peptides. Ideally, one could predict which y-ions will shift with the different location of a phosphorylation. Figure 4 illustrates such an example. The y8 ion is the only one predicted to differ between the two isoforms of singly-phosphorylated STFHAGQLR. The y82+ ion is observed in both spectra. Since this is not the case for all peptides, we considered several alternative strategies for differentiating between isoforms. FIGURE 1. Synthetic peptides and their modifications. Red letter indicates a phosphorylation site. A consolidated spectrum is built in the library when there are at least five spectra acquired for a peptide. Some peptides were observed at multiple charge states. FIGURE 4. Library spectra for two isoforms of peptide STFHAGQLR. Red S or T in the peptide sequence indicates the site of phosphrylation. Peaks are color-coded according to which isoforms they are common to. One peak is unique to each isoform. Some peaks predicted in both forms are only observed in one. All trademarks are the property of Thermo Fisher Scientific and its subsidiaries. This information is not intended to encourage use of these products in any manners that might infringe the intellectual property rights of others. peaks predicted and observed for both isoforms peaks predicted for both but observed in one isoform peaks predicted and observed in only one isoform