Download
1 / 1
20 likes | 156 Views
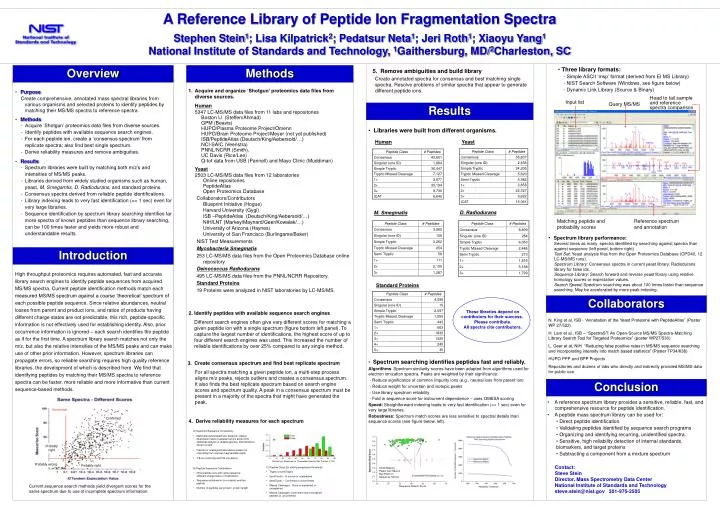
thresholds. Confirmed. Probably right. Probably wrong. Probably right. Head to tail sample and reference spectra comparison. Input list. Query MS/MS. Matching peptide and probability scores. Reference spectrum and annotation. A Reference Library of Peptide Ion Fragmentation Spectra

E N D
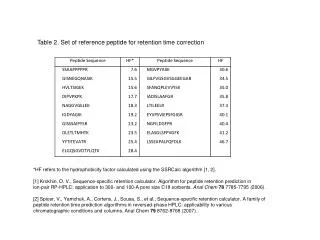
thresholds Confirmed Probably right Probably wrong Probably right Head to tail sample and reference spectra comparison Input list Query MS/MS Matching peptide and probability scores Reference spectrum and annotation A Reference Library of Peptide Ion Fragmentation Spectra Stephen Stein1; Lisa Kilpatrick2; Pedatsur Neta1; Jeri Roth1; Xiaoyu Yang1National Institute of Standards and Technology, 1Gaithersburg, MD/2Charleston, SC Overview Methods • Three library formats: • - Simple ASCII ‘msp’ format (derived from EI MS Library) • - NIST Search Software (Windows, see figure below) • - Dynamic Link Library (Source & Binary) • 5. Remove ambiguities and build library Create annotated spectra for consensus and best matching single spectra. Resolve problems of similar spectra that appear to generate different peptide ions. • Acquire and organize ‘Shotgun’ proteomics data files from diverse sources. • Human • 5347 LC-MS/MS data files from 11 labs and repositories • Boston U. (Steffen/Ahmad) • GPM (Beavis) • HUPO/Plasma Proteome Project/Omenn • HUPO/Brain Proteome Project/Meyer (not yet published) • ISB/PeptideAtlas (Deutsch/King/Aebersold/…) • NCI-SAIC (Veenstra) • PNNL/NCRR (Smith), • UC Davis (Rice/Lee) • Q-tof data from USB (Pannell) and Mayo Clinic (Muddiman) • Yeast • 2503 LC-MS/MS data files from 12 laboratories Online repositories PeptideAtlas Open Proteomics Database Collaborators/ContributorsBlueprint Initiative (Hogue)Harvard University (Gygi)ISB –PeptideAtlas (Deutsch/King/Aebersold/…)NIH/LNT (Markey/Maynard/Geer/Kowalak/…)University of Arizona (Haynes)University of San Francisco (Burlingame/Baker) NIST Test Measurements Mycobacteria Smegmatis 253 LC-MS/MS data files from the Open Proteomics Database online repository Deinococcus Radiodurans 495 LC-MS/MS data files from the PNNL/NCRR Repository. Standard Proteins 19 Proteins were analyzed in NIST laboratories by LC-MS/MS. • Purpose Create comprehensive, annotated mass spectral libraries from various organisms and selected proteins to identify peptides by matching their MS/MS spectra to reference spectra. • Methods - Acquire ‘Shotgun’ proteomics data files from diverse sources. - Identify peptides with available sequence search engines. • For each peptide ion, create a ‘consensus spectrum’ from replicate spectra; also find best single spectrum. • Derive reliability measures and remove ambiguities. • Results • Spectrum libraries were built by matching both m/z’s and intensities of MS/MS peaks. • Libraries derived from widely studied organisms such as human, yeast, M. Smegamtis, D. Radiodurans, and standard proteins. • Consensus spectra derived from reliable peptide identifications. • Library indexing leads to very fast identification (<< 1 sec) even for very large libraries. • Sequence identification by spectrum library searching identifies far more spectra of known peptides than sequence library searching, can be 100 times faster and yields more robust and understandable results. Results • Libraries were built from different organisms. Human Yeast D. Radiodurans M. Smegmatis • Spectrum library performance: • Several times as many spectra identified by searching against spectra than against sequence (left panel, bottom right) • Test Set: Yeast analysis files from the Open Proteomics Database (OPD40, 12 LC-MS/MS runs). • Spectrum Library: Consensus spectra in current yeast library. Radiodurans library for false ids. • Sequence Library: Search forward and reverse yeast library using relative homology scores or expectation values. • Search Speed: Spectrum searching was about 100 times faster than sequence searching. May be accelerated by more peak indexing. Introduction High throughput proteomics requires automated, fast and accurate library search engines to identify peptide sequences from acquired MS/MS spectra. Current peptide identification methods match each measured MS/MS spectrum against a coarse ‘theoretical’ spectrum of each possible peptide sequence. Since relative abundances, neutral losses from parent and product ions, and ratios of products having different charge states are not predictable, this rich, peptide-specific information is not effectively used for establishing identity. Also, prior occurrence information is ignored – each search identifies the peptide as if for the first time. A spectrum library search matches not only the m/z, but also the relative intensities of the MS/MS peaks and can make use of other prior information. However, spectrum libraries can propagate errors, so reliable searching requires high quality reference libraries, the development of which is described here. We find that identifying peptides by matching their MS/MS spectra to reference spectra can be faster, more reliable and more informative than current sequence-based methods. Standard Proteins Collaborators These libraries depend on contributors for their success. Please contribute. All spectra cite contributors. • 2. Identify peptides with available sequence search engines • Different search engines often give very different scores for matching a given peptide ion with a single spectrum (figure bottom left panel). To capture the largest number of identifications, the highest score of up to four different search engines was used. This increased the number of reliable identifications by over 25% compared to any single method. N. King et al, ISB - “Annotation of the Yeast Proteome with PeptideAtlas” (Poster WP 27/522) H. Lam et al., ISB – “SpectraST: An Open-Source MS/MS Spectra-Matching Library Search Tool for Targeted Proteomics” (poster WP27/530) L. Geer et al, NIH “Reducing false positive rates in MS/MS sequence searching and incorporating intensity into match based statistics” (Poster TP34/638) HUPO PPP and BPP Projects Repositories and dozens of labs who directly and indirectly provided MS/MS data for public use • 3. Create consensus spectrum and find best replicate spectrum • For all spectra matching a given peptide ion, a multi-step process aligns m/z peaks, rejects outliers and creates a consensus spectrum. It also finds the best replicate spectrum based on search engine scores and spectrum quality. A peak in a consensus spectrum must be present in a majority of the spectra that might have generated the peak. • Spectrum searching identifies peptides fast and reliably. • Algorithms: Spectrum similarity scores have been adapted from algorithms used for electron ionization spectra. Peaks are weighted by their significance: • - Reduce significance of common impurity ions (e.g., neutral loss from parent ion) • - Reduce weight for uncertain and isotopic peaks • - Use library spectrum reliability • - Fold in sequence score for instrument dependence – uses OMSSA scoring • Speed: Straightforward indexing leads to very fast identification (<< 1 sec) even for very large libraries. • Robustness: Spectrum match scores are less sensitive to spectral details than sequence scores (see figure below, left). Conclusion • A reference spectrum library provides a sensitive, reliable, fast, and comprehensive resource for peptide identification. • A peptide mass spectrum library can be used for: • Direct peptide identification • Validating peptides identified by sequence search programs • Organizing and identifying recurring, unidentified spectra. • Sensitive, high reliability detection of internal standards, biomarkers, and target proteins • Subtracting a component from a mixture spectrum 4. Derive reliability measures for each spectrum • A) Spectrum/Sequence Consistency • Match theoretical spectrum, based on relative dissociation rates of adjacent amino acids (from statistical analysis of reliable spectra). Discrimination shown at right • Fraction of unassigned abundance (peaks not originating from a known fragmentation path) • Y/B ion continuity and Y/B correlation Similarity of Measured vs. Theoretical Spectra (Dot Product x 100) Contact: Steve Stein Director, Mass Spectrometry Data Center National Institute of Standards and Technology steve.stein@nist.gov 301-975-2505 • C) Peptide Class (for setting acceptance threshold) • Tryptic or semiTryptic • SemiTryptic – In source or unexpected • SemiTryptic – Confirmed or unconfirmed • Missed Cleavages: None or explained, or unexplained • Missed Cleavages: Confirmed (found contained peptide) or unconfirmed Small Missing Peaks Can Have A Big Effect on Sequence Scores • B) Peptide Sequence Confirmation • Other peptide ions with same sequence (different charge state or modification) • Sequence contained in (or contains) another peptide • Number of peptides per protein / protein length Current sequence search methods yield divergent scores for the same spectrum due to use of incomplete spectrum information. Sequence Search Score