Download
1 / 18
190 likes | 313 Views
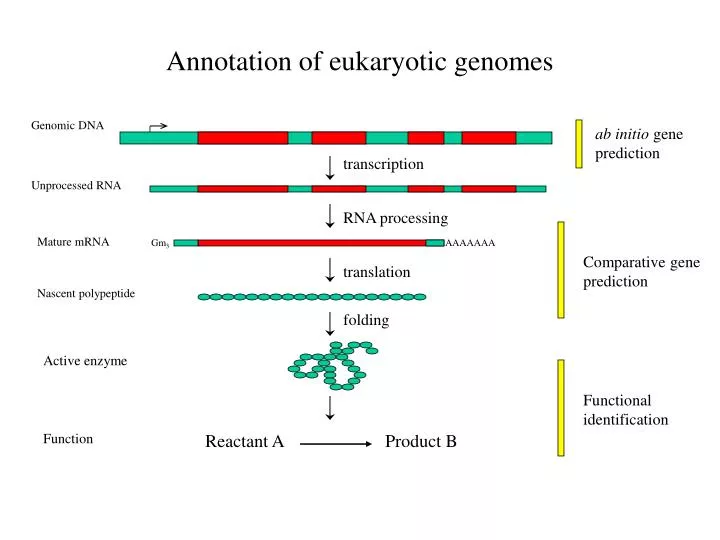
Annotation of eukaryotic genomes. Genomic DNA. ab initio gene prediction. transcription. Unprocessed RNA. RNA processing. Mature mRNA. Gm 3. AAAAAAA. Comparative gene prediction. translation. Nascent polypeptide. folding. Active enzyme. Functional identification. Function.

E N D
Annotation of eukaryotic genomes Genomic DNA ab initio gene prediction transcription Unprocessed RNA RNA processing Mature mRNA Gm3 AAAAAAA Comparative gene prediction translation Nascent polypeptide folding Active enzyme Functional identification Function Reactant A Product B
Gene finding: ab initio • What features of a ORF can we use? • Size - large open reading frames • DNA composition - codon usage / 3rd position codon bias • Other features: • Kozak sequence CCGCCAUGG • Ribosome binding sites • Termination signal (stops) • Splice junction boundaries
Gene finding: comparative • Use knowledge of known coding sequences to identify region of genomic DNA by similarity • transcribed DNA sequence • peptide sequence • related genomic sequence
Annotation of eukaryotic genomes Genomic DNA ab initio gene prediction transcription Unprocessed RNA RNA processing Mature mRNA Gm3 AAAAAAA Comparative gene prediction translation Nascent polypeptide folding Active enzyme Functional identification Function Reactant A Product B
Methods for searching • Pairwise alignments: matching a query sequence against a database of subject sequences • Needleman & Wunsch - global alignment • Smith-Waterman - local alignment • FastA • BLAST • Others: SSAHA, WABA • see Chapter 7 Developing Bionformatics Computer Skills
BLAST - local similarity searches • BLAST (Basic Local Alignment Search Tool) is the workhorse of genome annotation due to it’s early optimisation for the UNIX platform • Underlies most of the web-based servers world-wide • Comes in many flavours: • BLASTN - DNA against DNA • BLASTX - DNA against Protein • BLASTP - Protein against Protein • TBLASTN - Protein against DNA • TBLASTX - DNA against DNA at the peptide level
BLAST - results • BLAST returns high-scoring pairs (HSPs) with a score and p-value. Blast output files can be large and difficult to interpret. • Hence we need tools to make sense of the data - both to filter/process the file and to visualise the resulting multiple sequence alignments. • MSPcrunch - a post-processor for BLAST with a number of different output types. • BioPerl - modules for handling sequences and BLAST output
Standard similarity searches for first-pass annotation • genomic DNA v transcript data • BLASTN / EST_GENOME • TBLASTX • genomic DNA v genomic DNA • BLASTN • TBLASTX • genomic DNA v non-redundant protein data • BLASTX
Data for gene prediction • EST/mRNA - intra-species matches • TBLASTX - inter-species matches • BLASTX - intra-species matches • BLASTX - inter-species matches • Coding measures - genefinder, hexamer • Splice sites - consensus sequences
Manual review of gene predictions • Check concordance with transcript data • Check concordance with peptide similarity data • Check splice site usage (intron / exon boundaries) • Set of human appraised gene predictions. The translations of the CDS sequences are used for protein feature analysis and initial assignment (ID, function)