Download

1 / 1
10 likes | 122 Views
EDLIAYLK. MIFAGIK. TGPNLHGLFGR. Mass over charge ratio. Retention time. Comparison of base peak chromatograms. Implementation. Classical method. Bayesian method. Assumed peak retention time & range of inter experiments variability. Data. a priori law e.g retention time fluctuation.

E N D
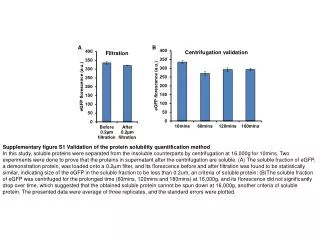
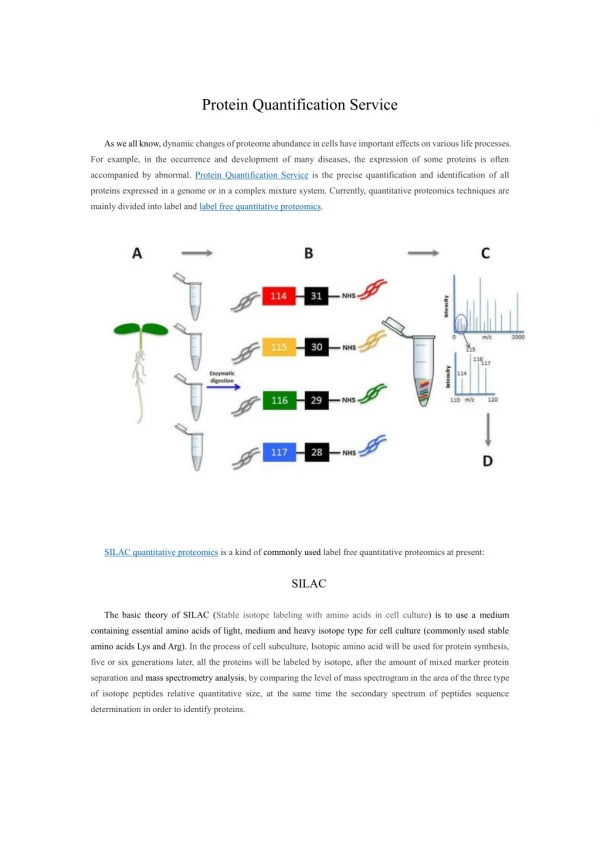
EDLIAYLK MIFAGIK TGPNLHGLFGR Mass over charge ratio Retention time Comparison of base peak chromatograms Implementation Classical method Bayesian method Assumed peak retention time & range of inter experiments variability Data a priori law e.g retention time fluctuation Isotopic patterns preprocessing m/z of peptides Data Signal model Data noise substraction Probabilistic bayesianmethod Iterative random generator Peak detection Peak integration Histogram of generated samples Parameter values (peak volume, retention time,noise level) Error margin Peak volume Robust protein quantification in mass spectrometry G.Strubel1, C.Paulus1, E. Mery1, L.Gerfault1, F. Ricoul1, V.Brun2, J.F. Giovannelli3, P.Grangeat1 1LETI, MINATEC, 2iRTSV,EDyP,CEA-GRENOBLE, 17 Rue des Martyrs, 38054 Grenoble Cedex 9, France 3Laboratoire des signaux et systèmes, UMR 8506 (CNRS-Supélec-UPS) Supélec, Gif-sur-Yvette, France gregory.strubel@cea.fr, laurent.gerfault@cea.fr Protein quantification based on LC-MS chain modeling and Bayesian inversion method • Introduction • Early detection of cancer by protein quantitation needs: sensitivity for early detection, selectivity to measure markers in complex mixture like plasma, accuracy and automatic measurement to allow screening. Moreover, experimental fluctuations need robust methods. The association of microfabricated column for liquid chromatography, mass spectrometer and dedicated signal processing will fulfil these needs. We propose a probabilistic Bayesian method based on acquisition system model to handle time retention fluctuation and peak overlapping. • Method characteristics • Probabilistic Bayesian method • Recover simultaneously peak volume, retention time, noise parameter • Combine simultaneously information of peptides of same protein to perform quantification • Consider all isotopic patterns & 2D peak volumes (fits two dimensions Gaussian shape on elution peaks) • Explicit noise model • Deliver values and error margin in respect to signal model used • Optional a priori information on parameters can be injected like parameter fluctuation range improving performances • Handle tagged peptides to deliver absolute quantification Handling of Peak overlapping 5 experiments performed with Cytochrome C digest in water buffer 3 concentrations: (0.2 µmol/L (3 exp.) , 0.5 µmol/L, 1 µmol/L. In the region of interest, 2 overlapped peaks of two peptides. No labelled peptides for absolute quantitation • Reproducible results for 0.2 µmol/L experiments • Concentration linearity recovered Quantification of pathogenic bacterial toxin (SEA)labelled with PSAQ in Urine The PSAQ quantification methodology [2] relies on the isotope dilution principle in which standardization is achieved with full-length isotope-labeled proteins. We compare the quantification obtained manually, from nano-LC-MS data and proposed Bayesian approach. In both case, data are preprocessed to subtract chemical noise appearing as continuous stripes in time direction and regularly spaced in mass over ratio direction. For manual quantification, an extra data smoothing filter is applied. For Bayesian method, the same set of parameters are used for all the experiments. The typical computation time is less than 10 seconds from preprocessed extracted data. For considered peptide, the relative retention time variation is 3%. Comparison of extractedchromatogram and spectrogramand the corresponding simulated ones from estimated parameters Comparison of quantification of SEA toxin in urineby manual and Bayesian methods Results The results obtained are similar to the ones obtained with manual method. The average variation coefficient is 6.2 %. This coefficient takes into account all sources of variations (experimental, instrumental and measurement). The comparison of measured and estimated signals shows that we are able to explain the signal content and control the sources of variation (mainly instrumental) included in the signal model. • Conclusions • We present a Bayesian approach dedicated to protein quantification. This automatic algorithm delivers results similar to the manual quantification obtained with extra preprocessing for complex samples such as urine. • We have also demonstrated its ability to quantify two fully overlapped peaks. • Nevertheless, quantification on SEA toxin has been performed for one peptide not affected by overlapping and with good signal to noise ratio. The proposed method is attended to overpass classical method performances for more complex mixtures. • References • (1)Bayesian estimation for molecular profile reconstruction in proteomics based on liquid chromatography and mass spectrometry, G. Strubel, et al. in EMBS '07. 29th Annual International Conference of the IEEE EMBS Lyon, August 2007. • (2) Isotope-labeled protein standards: Towards absolute quantitative proteomics, Brun V,et al.,Mol Cell Proteomics. 2007 Dec;6(12):2139-49. • (3) Lab-On-Chip based protein profiling for CANcer DIAgnosis project(http://www.loccandia.eu)